UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| ☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | ||||
For the fiscal year ended December 31, 2020
OR
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | ||||
For the transition period from to
Commission File Number 001-32335
___________________________
HALOZYME THERAPEUTICS, INC.
(Exact name of registrant as specified in its charter)
___________________________
| Delaware | 88-0488686 | |||||||
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) | |||||||
| 11388 Sorrento Valley Road | 92121 | |||||||
| San Diego | (Zip Code) | |||||||
| CA | ||||||||
| (Address of principal executive offices) | ||||||||
(858) 794-8889
(Registrant’s telephone number, including area code)
Securities registered under Section 12(b) of the Act:
| Title of Each Class | Trading Symbol(s) | Name of Each Exchange on Which Registered | ||||||
| Common Stock, $0.001 Par Value | HALO | The NASDAQ Stock Market, LLC | ||||||
Securities registered under Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. ☒ Yes ☐ No
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. ☐ Yes ☒ No
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. ☒ Yes ☐ No
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). ☒ Yes ☐ No
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer”, “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | Accelerated filer | Non-accelerated filer | Smaller reporting company | Emerging growth company | |||||||||||||
| ☒ | ☐ | ☐ | ☐ | ☐ | |||||||||||||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐ | |||||||||||||||||
Indicate by check mark whether the Registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☒
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). ☐ Yes ☒ No
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant as of June 30, 2020 was approximately $2.9 billion based on the closing price on the NASDAQ Global Select Market reported for such date. Shares of common stock held by each officer and director and by each person who is known to own 10% or more of the outstanding common stock have been excluded in that such persons may be deemed to be affiliates of the registrant. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
The number of outstanding shares of the registrant’s common stock, par value $0.001 per share, was 135,276,380 as of February 17, 2021.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive Proxy Statement to be filed subsequent to the date hereof with the Securities and Exchange Commission pursuant to Regulation 14A in connection with the registrant’s 2021 Annual Meeting of Stockholders are incorporated by reference into Part III of this Annual Report.
HALOZYME THERAPEUTICS, INC.
INDEX
| Page | |||||||||||
| Item 1. | |||||||||||
| Item 1A. | |||||||||||
| Item 1B. | |||||||||||
| Item 2. | |||||||||||
| Item 3. | |||||||||||
| Item 4. | |||||||||||
| Item 5. | |||||||||||
| Item 6. | |||||||||||
| Item 7. | |||||||||||
| Item 7A. | |||||||||||
| Item 8. | |||||||||||
| Item 9. | |||||||||||
| Item 9A. | |||||||||||
| Item 9B. | |||||||||||
| Item 10. | |||||||||||
| Item 11. | |||||||||||
| Item 12. | |||||||||||
| Item 13. | |||||||||||
| Item 14. | |||||||||||
| Item 15. | |||||||||||
| Item 16. | |||||||||||
This Annual Report on Form 10-K contains “forward-looking statements” within the meaning of the “safe harbor” provisions of Section 21E of the Securities Exchange Act of 1934, as amended, and Section 27A of the Securities Act of 1933, as amended. All statements, other than statements of historical fact, included herein, including without limitation those regarding our future product development and regulatory events and goals, product collaborations, our business intentions and financial estimates and anticipated results, are, or may be deemed to be, forward-looking statements. Words such as “expect,” “anticipate,” “intend,” “plan,” “believe,” “seek,” “estimate,” “think,” “may,” “could,” “will,” “would,” “should,” “continue,” “potential,” “likely,” “opportunity,” “project” and similar expressions or variations of such words are intended to identify forward-looking statements, but are not the exclusive means of identifying forward-looking statements in this Annual Report. Additionally, statements concerning future matters such as the development or regulatory approval of new partner products, enhancements of existing products or technologies, timing and success of launch of new products by our collaborators, third party performance under key collaboration agreements, revenue, expense, profitability and cash flow levels, and expected trends and other statements regarding matters that are not historical are forward-looking statements.
Although forward-looking statements in this Annual Report reflect the good faith judgment of our management, such statements can only be based on facts and factors currently known by us. Consequently, forward-looking statements are inherently subject to risks and uncertainties and actual results and outcomes may differ materially from the results and outcomes discussed in or anticipated by the forward-looking statements. Factors that could cause or contribute to such differences in results and outcomes include without limitation those discussed under the heading “Risk Factors” in Part I, Item 1A below, as well as those discussed elsewhere in this Annual Report. Readers are urged not to place undue reliance on these forward-looking statements, which speak only as of the date of this Annual Report. We undertake no obligation to revise or update any forward-looking statements in order to reflect any event or circumstance that may arise after the date of this Annual Report. Readers are urged to carefully review and consider the various disclosures made in this Annual Report, which attempt to advise interested parties of the risks and factors that may affect our business, financial condition, results of operations and prospects.
References to “Halozyme,” “the Company,” “we,” “us,” and “our” refer to Halozyme Therapeutics, Inc. and its wholly owned subsidiary, Halozyme, Inc., and Halozyme, Inc.’s wholly owned subsidiaries, Halozyme Holdings Ltd., Halozyme Switzerland GmbH and Halozyme Switzerland Holdings GmbH. References to “Notes” refer to the Notes to Consolidated Financial Statements included herein (refer to Part II, Item 8).
PART I
1
Item 1.Business
Overview
Halozyme Therapeutics Inc. is a biopharma technology platform company that provides innovative and disruptive solutions with the goal of improving patient experience and outcomes. Our proprietary enzyme, rHuPH20, is used to facilitate the delivery of injected drugs and fluids. We license our technology to biopharmaceutical companies to collaboratively develop products that combine our ENHANZE® drug delivery technology with the collaborators’ proprietary compounds.
Our approved product and our collaborators’ approved products and product candidates are based on rHuPH20, our patented recombinant human hyaluronidase enzyme. rHuPH20 is the active ingredient in our first commercially approved product, Hylenex® recombinant (Hylenex), and it works by breaking down hyaluronan (or HA), a naturally occurring carbohydrate that is a major component of the extracellular matrix in tissues throughout the body such as skin and cartilage. This temporarily increases dispersion and absorption allowing for improved subcutaneous delivery of injectable biologics, such as monoclonal antibodies and other large therapeutic molecules, as well as small molecules and fluids. We refer to the application of rHuPH20 to facilitate the delivery of other drugs or fluids as our ENHANZE® drug delivery technology (ENHANZE). We license the ENHANZE technology to form collaborations with biopharmaceutical companies that develop or market drugs requiring or benefiting from injection via the subcutaneous route of administration. In the development of proprietary intravenous (IV) drugs combined with our ENHANZE technology, data have been generated supporting the potential for ENHANZE to reduce treatment burden, as a result of shorter duration of subcutaneous (SC) administration. ENHANZE may enable fixed-dose SC dosing compared to weight-based dosing required for IV administration, and potentially allow for lower rates of infusion related reactions. ENHANZE may enable more flexible treatment options such as home administration by a healthcare professional or potentially the patient. Lastly, certain proprietary drugs co-formulated with ENHANZE have been granted additional exclusivity, extending the patent life of the product beyond the one of the proprietary IV drug.
We currently have ENHANZE collaborations with F. Hoffmann-La Roche, Ltd. and Hoffmann-La Roche, Inc. (Roche), Baxalta US Inc. and Baxalta GmbH (now members of the Takeda group of companies, following the acquisition of Shire plc by Takeda Pharmaceutical Company Limited in January 2019) (Baxalta), Pfizer Inc. (Pfizer), Janssen Biotech, Inc. (Janssen), AbbVie, Inc. (AbbVie), Eli Lilly and Company (Lilly), Bristol-Myers Squibb Company (BMS), Alexion Pharma Holding (Alexion), ARGENX BVBA (argenx) and Horizon Therapeutics plc. (Horizon). We receive royalties from three of these collaborations, including royalties from sales of one product from the Baxalta collaboration, three products from the Roche collaboration and one product from the Janssen collaboration. Future potential revenues from royalties and fees from ENHANZE collaborations and the sales and/or royalties of our approved products will depend on the ability of Halozyme and our collaborators to develop, manufacture, secure and maintain regulatory approvals for approved products and product candidates and commercialize product candidates.
Our principal offices and research facilities are located at 11388 Sorrento Valley Road, San Diego, California 92121. Our telephone number is (858) 794-8889 and our e-mail address is info@halozyme.com. Our website address is www.halozyme.com. Information found on, or accessible through, our website is not a part of, and is not incorporated into, this Annual Report on Form 10-K. Our periodic and current reports that we filed with the SEC are available on our website at www.halozyme.com, free of charge, as soon as reasonably practicable after we have electronically filed such material with, or furnished them to, the SEC, including our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and any amendments to those reports.
Technology
rHuPH20 can be applied as a drug delivery platform to increase dispersion and absorption of other injected drugs and fluids potentially reducing treatment burden. For example, rHuPH20 has been used to convert drugs that must be delivered intravenously into subcutaneous injections or to reduce the number of subcutaneous injections needed for effective therapy. When ENHANZE technology is applied subcutaneously, the rHuPH20 acts locally and transiently, with a tissue half-life of less than 15 minutes. HA at the local site reconstitutes its normal density within a few days and, therefore, the effect of rHuPH20 on the architecture of the subcutaneous space is temporary.
2
Strategy
We are a leader in converting IV biologics to subcutaneous delivery using our commercially-validated ENHANZE technology. We collaborate with leading pharmaceutical and biotechnology companies to help them develop products that combine our ENHANZE technology with their proprietary compounds. We target large, attractive markets, where ENHANZE-enabled subcutaneous delivery has the potential to deliver competitive differentiation and other important benefits to our partners, such as larger injection volumes administered rapidly, extended dosing intervals, and reduced treatment burden and healthcare costs. In addition, ENHANZE has been demonstrated to enable the combination of two therapeutic antibodies in a single injection, as well as the development of new co-formulation intellectual property. We leverage our strategic, technical, regulatory and alliance management skills in support of our partners' efforts to develop new subcutaneously delivered products. We currently have ten collaborations with five current product approvals and additional product candidates in development using our ENHANZE technology. We intend to work with our existing collaborators to expand our collaborations to add new targets and develop targets and product candidates under the terms of the operative collaboration agreements. We will also continue our efforts to enter into new collaborations to further derive additional value from our proprietary technology.
3
Product and Product Candidates
We currently have one marketed proprietary product and five marketed partnered products. The following table summarizes our proprietary product, marketed partnered products and product candidates under development with our collaborators:
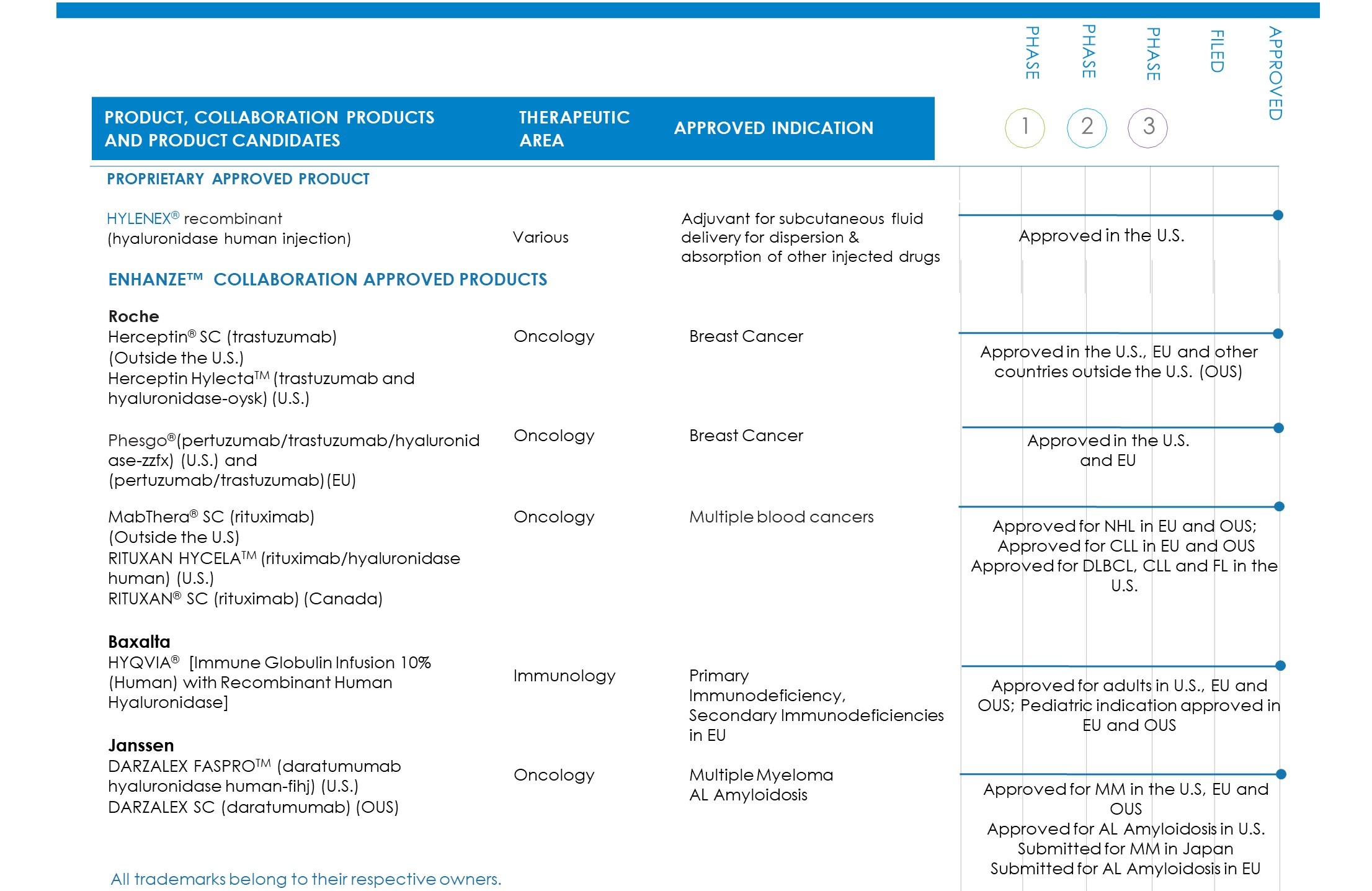
4
Proprietary Product
Hylenex Recombinant (hyaluronidase human injection)
Hylenex recombinant is a formulation of rHuPH20 that facilitates subcutaneous fluid administration for achieving hydration, to increase the dispersion and absorption of other injected drugs and, in subcutaneous urography, to improve resorption of radiopaque agents. Hylenex recombinant is currently the number one prescribed branded hyaluronidase.
ENHANZE Collaborations
Roche Collaboration
In December 2006, we and Roche entered into a collaboration and license agreement under which Roche obtained a worldwide license to develop and commercialize product combinations of rHuPH20 and up to thirteen Roche target compounds (the Roche Collaboration). Under this agreement, Roche elected a total of eight targets, two of which are exclusive.
In September 2013, Roche launched a subcutaneous (SC) formulation of Herceptin (trastuzumab) (Herceptin SC) in Europe for the treatment of patients with HER2-positive breast cancer followed by launches in additional countries. This formulation utilizes our ENHANZE technology and is administered in two to five minutes, compared to 30 to 90 minutes with the standard intravenous form. In September 2018, we announced that Roche received approval from Health Canada for Herceptin SC for the treatment of patients with HER2-positive breast cancer. In February 2019, we announced that Roche received approval from the U.S. Food and Drug Administration (FDA) for Herceptin SC under the brand name Herceptin Hylecta™ (trastuzumab and hyaluronidase-oysk). In April 2019, Roche made Herceptin Hylecta available in the U.S.
Directed at the same target, Roche initiated a Phase 1 study of Perjeta® (pertuzumab) and Herceptin (trastuzumab) using ENHANZE technology in patients with early breast cancer in March 2016. In June 2018, Roche initiated a global Phase 3 study of a fixed-dose combination of Perjeta and Herceptin using ENHANZE technology in patients with HER2-positive early breast cancer. In August 2019, the global phase 3 study met its primary endpoint. The study results demonstrated non-inferior levels of Perjeta in the blood (pharmacokinetics) compared to standard intravenous (IV) infusion of Perjeta plus Herceptin and chemotherapy in patients with HER2-positive early breast cancer. The study also demonstrated that the safety profile of the fixed dose subcutaneous combination of Perjeta and Herceptin was consistent with the safety profile of Perjeta and Herceptin administered intravenously. In June 2020, the FDA approved the fixed-dose combination of Perjeta and Herceptin for subcutaneous injection utilizing ENHANZE technology (Phesgo®) for the treatment of patients with HER2-positive breast cancer. In December 2020, the European Commission (EC) also approved Phesgo for the treatment of patients with early and metastatic HER2-positive breast cancer.
In June 2014, Roche launched MabThera SC in Europe for the treatment of patients with common forms of non-Hodgkin lymphoma (NHL) followed by launches in additional countries. This formulation utilizes our ENHANZE technology and is administered in approximately five minutes compared to the approximately 1.5 to 4 hour intravenous infusion. In May 2016, Roche announced that the European Medicines Agency (EMA) approved Mabthera SC to treat patients with chronic lymphocytic leukemia (CLL). In June 2017, the FDA approved Genentech’s RITUXAN HYCELA™, a combination of rituximab using ENHANZE technology (approved and marketed under the MabThera SC brand in countries outside the U.S. and Canada), for CLL and two types of NHL, follicular lymphoma and diffuse large B-cell lymphoma. In March 2018, Health Canada approved a combination of rituximab and rHuPH20 (approved and marketed under the brand name RITUXAN® SC) for patients with CLL.
In September 2017, we and Roche entered into an agreement providing Roche the right to develop and commercialize one additional exclusive target using ENHANZE technology. The upfront license payment may be followed by event-based payments subject to Roche’s achievement of specified development, regulatory and sales-based milestones. In addition, Roche will pay royalties to us if products under the collaboration are commercialized.
In October 2018, we entered into an agreement with Roche for the right to develop and commercialize one additional exclusive target and an option to select two additional targets within four years using ENHANZE technology. The upfront license payment may be followed by event-based payments subject to Roche’s achievement of specified development, regulatory and sales-based milestones. In addition, Roche will pay royalties to us if products under the collaboration are commercialized.
5
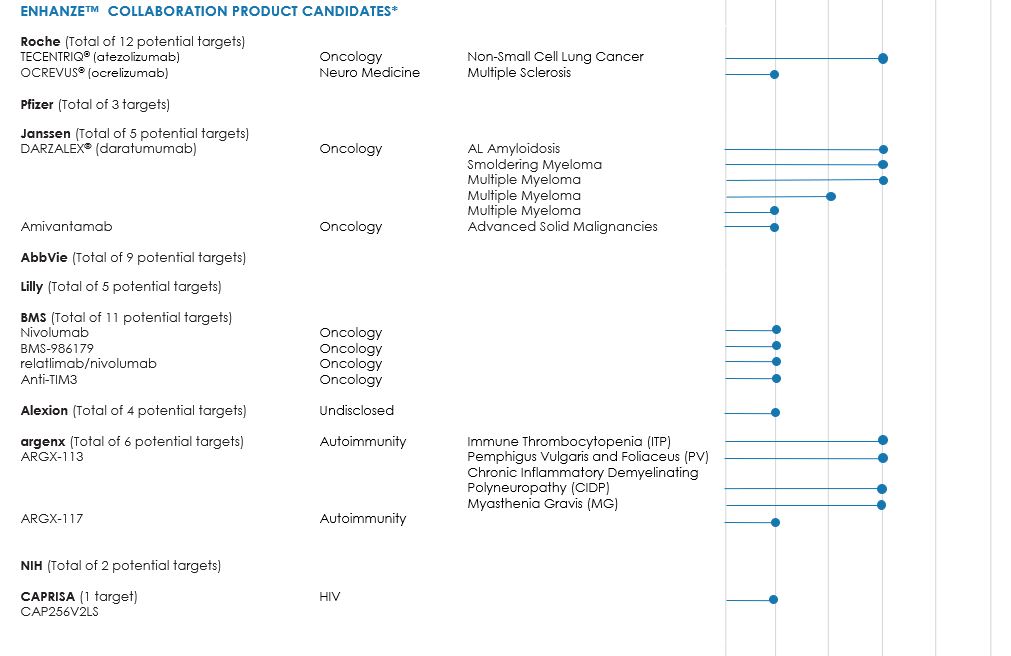
In December 2018, Roche initiated a Phase 1b/2 study in patients with non-small cell lung cancer for Tecentriq (atezolizumab) using ENHANZE technology. In September 2020, Roche presented a poster with data from Part 1 of its Phase 1b study (IMscin001) evaluating atezolizumab (Tecentriq) for subcutaneous administration utilizing ENHANZE technology in patients with locally advanced or metastatic non-small cell lung cancer at the ESMO Virtual Congress 2020. The poster concluded that atezolizumab utilizing ENHANZE technology provided similar exposure as atezolizumab IV and that results support further development of subcutaneous atezolizumab in IMscin001 Part 2, a confirmatory Phase 3 study. In December 2020, Roche initiated a Phase 3 study in patients with non-small cell lung cancer for Tecentriq using ENHANZE technology.
In August 2019, Roche initiated a Phase 1 study evaluating OCREVUS (ocrelizumab) with ENHANZE technology in subjects with multiple sclerosis.
In October 2019, Roche nominated a new undisclosed target to be studied using ENHANZE technology, triggering a $10 million milestone payment.
Baxalta Collaboration
In September 2007, we and Baxalta entered into a collaboration and license agreement under which Baxalta obtained a worldwide, exclusive license to develop and commercialize product combinations of rHuPH20 with GAMMAGARD LIQUID (HYQVIA) (the Baxalta Collaboration). HYQVIA is indicated for the treatment of primary immunodeficiency disorders associated with defects in the immune system.
In May 2013, the European Commission granted Baxalta marketing authorization in all EU Member States for the use of HYQVIA (solution for subcutaneous use) as replacement therapy for adult patients with primary and secondary immunodeficiencies. Baxalta launched HYQVIA in the first EU country in July 2013 and has continued to launch in additional countries.
In September 2014, HYQVIA was approved by the FDA for treatment of adult patients with primary immunodeficiency in the U.S. HYQVIA is the first subcutaneous immune globulin (IG) treatment approved for adult primary immunodeficiency patients with a dosing regimen requiring only one infusion up to once per month (every three to four weeks) and one injection site per infusion in most patients, to deliver a full therapeutic dose of IG. The FDA’s approval of HYQVIA was a significant milestone for us as it represented the first U.S. approved BLA which utilizes our rHuPH20 platform.
In May 2016, Baxalta announced that HYQVIA received a marketing authorization from the European Commission for a pediatric indication, which was launched in Europe to treat primary and certain secondary immunodeficiencies. In September 2020, Takeda announced that the EMA approved a label update for HYQVIA broadening its use and making it the first and only facilitated subcutaneous immunoglobulin replacement therapy in adults, adolescents and children with an expanded range of secondary immunodeficiencies (SID).
Pfizer Collaboration
In December 2012, we and Pfizer entered into a collaboration and license agreement, under which Pfizer has the worldwide license to develop and commercialize products combining our rHuPH20 enzyme with Pfizer proprietary biologics in primary care and specialty care indications. Pfizer has elected five targets and has returned two targets.
Janssen Collaboration
In December 2014, we and Janssen entered into a collaboration and license agreement, under which Janssen has the worldwide license to develop and commercialize products combining our rHuPH20 enzyme with Janssen proprietary biologics directed to up to five targets. Targets may be selected on an exclusive basis. Janssen has elected CD38 as the first target on an exclusive basis. Janssen has initiated seven Phase 3 studies, two Phase 2 study and one Phase 1 study of DARZALEX® (daratumumab), directed at CD38, using ENHANZE technology in patients with amyloidosis, smoldering myeloma and multiple myeloma.
In February 2019, Janssen’s development partner, Genmab, announced positive Phase 3 trial results from the COLUMBA study evaluating subcutaneous DARZALEX in comparison to DARZALEX IV in patients with relapsed or refractory multiple myeloma. DARZALEX SC (utilizing ENHANZE technology) was found to be non-inferior to Darzalex IV with regard the co-primary endpoints of Overall Response Rate and Maximum Trough concentration. In May 2020, we announced that Janssen received US FDA approval and launched the commercial sale of DARZALEX FASPRO in four regimens across five indications in multiple myeloma patients, including newly diagnosed, transplant-ineligible patients as well as relapsed or refractory patients. As a fixed-dose formulation, DARZALEX FASPRO can be administered over three to five minutes, significantly less time than IV DARZALEX which requires multi-hour infusions. In June 2020, we announced that Janssen received European marketing authorization and launched the commercial sale of DARZALEX SC utilizing ENHANZE in the European Union. In April 2020, we announced the submission of a New Drug Application (NDA) to Japan's Ministry of Health, Labour and Welfare (MHLW) by Janssen seeking approval of DARZALEX SC. In November 2020, Janssen announced it submitted a Type II variation application to the EMA seeking European approval of DARZALEX SC to be used in the
6
treatment of patients with AL amyloidosis, a rare and potentially fatal disease for which there are no currently approved therapies. In August 2020, Janssen announced that Health Canada approved DARZALEX SC in four regimens across five indications in patients with multiple myeloma, most notably newly diagnosed, trans-plant-ineligible patients as well as relapsed or refractory patients. In November 2020, Janssen filed for approval of DARZALEX FASPRO/Darzalex SC with pomalidomide and dexamethasone for patients with relapsed or refractory multiple myeloma in the U.S. and EU. In January 2021, Janssen received FDA approval for DARZALEX FASPRO in combination with bortezomib, thalidomide, and dexamethasone in newly diagnosed multiple myeloma patients who are eligible for autologous stem cell transplant. In January 2021, Janssen received accelerated approval from the FDA for DARZALEX FASPRO in combination with bortezomib, cyclophosphamide and dexamethasone (D-VCd) for the treatment of adult patients with newly diagnosed AL amyloidosis (not recommended for the treatment of patients with AL amyloidosis who have NYHA Class IIIB or Class IV cardiac disease or Mayo Stage IIIB outside of controlled clinical trials).
In December 2019, Janssen elected targets EGFR and cMET on an exclusive basis as part of the bispecific antibody (amivantamab), which is being studied in solid tumors. In November 2020, Janssen initiated a Phase 1 study of amivantamab and ENHANZE.
AbbVie Collaboration
In June 2015, we and AbbVie entered into a collaboration and license agreement, under which AbbVie has the worldwide license to develop and commercialize products combining our rHuPH20 enzyme with AbbVie proprietary biologics directed to up to nine targets. Targets may be selected on an exclusive basis. AbbVie elected one target on an exclusive basis, TNF alpha, for which it has discontinued development and returned the target.
Lilly Collaboration
In December 2015, we and Lilly entered into a collaboration and license agreement, under which Lilly has the worldwide license to develop and commercialize products combining our rHuPH20 enzyme with Lilly proprietary biologics directed to up to five targets. Targets may be selected on an exclusive basis. Lilly has elected two targets on an exclusive basis and one target on a semi-exclusive basis. In August 2017, Lilly initiated a Phase 1 study of an investigational therapy in combination with rHuPH20.
BMS Collaboration
In September 2017, we and BMS entered into a collaboration and license agreement, which became effective in November 2017, under which BMS has the worldwide license to develop and commercialize products combining our rHuPH20 enzyme with BMS products directed at up to eleven targets. Targets may be selected on an exclusive basis. BMS has designated multiple immuno-oncology targets including programmed death 1 (PD-1) and has an option to select additional targets within five years from the effective date. In October 2018, BMS dosed the first patient in a Phase 1/2a study evaluating the safety, pharmacokinetics and pharmacodynamics of BMS-986179, an investigational anti-CD-73 antibody alone and in combination with nivolumab, using ENHANZE technology. BMS is also conducting a Phase 1/2 study of nivolumab using ENHANZE technology in patients with solid tumors. In October 2019, BMS initiated a Phase 1 study for relatlimab in combination with nivolumab using ENHANZE technology. In June 2020, BMS initiated a Phase 1/2 study of ipilimumab in combination with nivolumab using ENHANZE technology, but has recently made a portfolio prioritization decision to not continue the study. In June 2020, BMS selected 3 targets on an exclusive basis and exercised their option to convert a co-exclusive license to an exclusive license. BMS has selected eight targets on an exclusive basis to-date.
Alexion Collaboration
In December 2017, we and Alexion entered into a collaboration and license agreement, under which Alexion has the worldwide license to develop and commercialize products combining our rHuPH20 enzyme with Alexion’s portfolio of products directed at up to four targets. Targets may be selected on an exclusive basis. Alexion elected two targets on an exclusive basis, including a C5 complement inhibitor and has an option to select two additional targets within five years from the effective date. In August 2018, Alexion announced that it initiated a Phase 1 trial to study a next-generation subcutaneous formulation of ALXN1210 using ENHANZE technology (ALXN1810). In April 2020 Alexion announced it would no longer proceed with a study of ALXN1810 in renal disease. Following a portfolio prioritization, Alexion has made the decision to not proceed with further development of ALXN1810, subcutaneous ULTOMIRIS co-administered with ENHANZE technology. Alexion is evaluating additional development options for ENHANZE.
7
argenx Collaboration
In February 2019, we and argenx entered into an agreement for the right to develop and commercialize one exclusive target, the human neonatal Fc receptor FcRn, which includes argenx's lead asset efgartigimod (ARGX-113), and an option to select two additional targets using ENHANZE technology. In May 2019, argenx nominated a second target to be studied using ENHANZE technology, a human complement factor C2 associated with the product candidate ARGX-117, which is being developed to treat severe autoimmune diseases.
In July 2019, argenx dosed the first subject in a phase 1 clinical trial evaluating the safety, pharmacokinetics and pharmacodynamics of efgartigimod (ARGX-113), using ENHANZE technology. In December 2019, argenx reported that based on data from the phase 1 study and internal company analysis, a one minute injection administered every 2 weeks may be possible. In December 2020, argenx initiated a Phase 3 study of ARGX-113 using ENHANZE technology for patients with immune thrombocytopenia (ITP), an immune disorder in which the blood does not clot normally. In January 2021, argenx initiated a Phase 3 study of ARGX-113 using ENHANZE technology in pemphigus vulgaris and foliaceus (PV), a rare autoimmune disease that causes painful blisters on the skin and mucous membranes. In February 2021, argenx initiated a Phase 3 study of ARGX-113 using ENHANZE technology for patients with chronic inflammatory demyelinating polyneuropathy (CIDP) and initiated a Phase 3 study of ARGX-113 using ENHANZE technology in myasthenia gravis (MG), an autoimmune disorder of the musculoskeletal system caused by IgG autoantibodies.
In October 2020, we and argenx entered into agreement to expand the collaboration relationship. Under the newly announced expansion, argenx gained the ability to exclusively access our ENHANZE technology for three additional targets upon nomination for a total of up to six targets under the existing and newly expanded collaboration.
Horizon Collaboration
In November 2020, we and Horizon entered into a global collaboration and license agreement that gives Horizon exclusive access to Halozyme's ENHANZE technology for subcutaneous formulation of medicines targeting IGF-1R. Horizon intends to use ENHANZE to develop a SC formulation of TEPEZZA (teprotumumab-trbw), indicated for the treatment of thyroid eye disease, a serious, progressive and vision-threatening rare autoimmune disease, potentially shortening drug administration time, reducing healthcare practitioner time and offering additional flexibility and convenience for patients.
NIH CRADA
In June 2019, we announced a Cooperative Research and Development Agreement (CRADA) with the National Institute of Allergy and Infectious Diseases’ Vaccine Research Center (VRC), part of National Institute of Health (NIH), enabling the VRC’s use of ENHANZE technology to develop subcutaneous formulations of broadly neutralizing antibodies (bnAbs) against HIV for HIV treatment. The initiation of this study has been impacted by COVID-19 and is delayed.
CAPRISA
In September 2020, we entered into a collaboration with the Centre for the AIDS Programme of Research in South Africa (CAPRISA), a non-profit company, to evaluate safety, tolerability and pharmacokinetics of a human monoclonal antibody (CAP256V2LS) in HIV-negative and HIV-positive women in South Africa. In October 2020, we were notified that the first patient was dosed with PH20 in a Phase I Dose-Escalation Study for CAP256V2LS.
For a further discussion of the collaboration agreements, refer to Note 2, Summary of Significant Accounting Policies - Revenues under Collaborative Agreements.
Impact of COVID-19 to our Business
In March 2020, the World Health Organization declared a disease caused by a strain of novel coronavirus (“COVID-19”) to be a pandemic. In an effort to contain COVID-19 or slow its spread, governments around the world have enacted various measures, including orders to close all businesses not deemed “essential,” isolate residents to their homes or places of residence, and practice social distancing when engaging in essential activities. In an effort to protect the health and safety of our employees and in compliance with state regulations, we instituted working from home, limited the number of people that work on site at any one time, and suspended employee travel. We anticipate that the global health crisis caused by COVID-19 will continue to have an impact across the globe. As an organization we continue to effectively operate with the majority of our employees working from home and not traveling, which we expect will continue for the foreseeable future. Importantly, our suppliers continue to operate without interruption related to COVID-19. However, the duration of the pandemic and its continued impact on the global economy as a whole is unknown at this time. We are not clear the extent to which near term and long term operational and economic impacts of COVID-19, if any, will have on our business, including the effects on our suppliers, collaborators, customers, employees, and prospects. We will continue to monitor the COVID-19 situation closely.
8
Patents and Proprietary Rights
Patents and other proprietary rights are essential to our business. Our success will depend in part on our ability to obtain patent protection for our inventions, to preserve our trade secrets and to operate without infringing the proprietary rights of third parties. Our strategy is to actively pursue patent protection in the U.S. and certain foreign jurisdictions for technology that we believe to be proprietary to us and that offers us a potential competitive advantage. Our patent portfolio includes 43 issued patents in the U.S., more than 460 issued patents in Europe and other countries in the world and more than 50 pending patent applications. In general, patents have a term of 20 years from the application filing date or earlier claimed priority date. Our issued patents will expire between 2023 and 2035. While we recently abandoned a number of patents related to PEGPH20 as a result of its failed Phase 3 clinical trial for the treatment of pancreatic cancer, we continue to file and prosecute patent applications to strengthen and grow our patent portfolio pertaining to our recombinant human hyaluronidase. We have multiple patents and patent applications throughout the world pertaining to our recombinant human hyaluronidase and methods of use and manufacture, including an issued U.S. patent which expires in 2027 and an issued European patent which expires in 2024, which we believe cover the products and product candidates under our existing collaborations and Hylenex recombinant. In addition, we have, under prosecution throughout the world, multiple patent applications that relate specifically to individual product candidates under development, the expiration of which can only be definitely determined upon maturation into our issued patents. We believe our patent filings represent a barrier to entry for potential competitors looking to utilize these hyaluronidases.
In addition to patents, we rely on unpatented trade secrets, proprietary know-how and continuing technological innovation. We seek protection of these trade secrets, proprietary know-how and innovation, in part, through confidentiality and proprietary information agreements. Our policy is to require our employees, directors, consultants, advisors, collaborators, outside scientific collaborators and sponsored researchers, other advisors and other individuals and entities to execute confidentiality agreements upon the start of employment, consulting or other contractual relationships with us. These agreements provide that all confidential information developed or made known to the individual or entity during the course of the relationship is to be kept confidential and not disclosed to third parties except in specific circumstances. In the case of employees and some other parties, the agreements provide that all inventions conceived by the individual will be our exclusive property. Despite the use of these agreements and our efforts to protect our intellectual property, there will always be a risk of unauthorized use or disclosure of information. Furthermore, our trade secrets may otherwise become known to, or be independently developed by, our competitors.
We also file trademark applications to protect the names of our products and product candidates. These applications may not mature to registration and may be challenged by third parties. We are pursuing trademark protection in a number of different countries around the world. There can be no assurances that our registered or unregistered trademarks or trade names will not infringe on rights of third parties or will be acceptable to regulatory agencies.
Research and Development Activities
Our research and development expenses consist primarily of costs associated with the product development, quality and regulatory work required to maintain the ENHANZE platform, development and manufacturing of product candidates performed on behalf of our partners, compensation and other expenses for research and development personnel, supplies and materials, facility costs and amortization and depreciation. We charge all research and development expenses to operations as they are incurred. Prior to our November 2019 restructuring, our research and development activities were primarily focused on the development of PEGPH20.
Manufacturing
We do not have our own manufacturing facility for our product and our partners’ products and product candidates, or the capability to package our products. We have engaged third parties to manufacture bulk rHuPH20 and Hylenex.
We have existing supply agreements with contract manufacturing organizations Avid Bioservices, Inc. (Avid) and Catalent Indiana LLC (Catalent) to produce supplies of bulk rHuPH20. These manufacturers each produce bulk rHuPH20 under current Good Manufacturing Practices (cGMP) for clinical and commercial uses. Catalent currently produces bulk rHuPH20 for use in Hylenex and collaboration product candidates. Avid currently produces bulk rHuPH20 for use in collaboration products. We rely on their ability to successfully manufacture these batches according to product specifications. It is important for our business for Catalent and Avid to (i) retain their status as cGMP-approved manufacturing facilities; (ii) successfully scale up bulk rHuPH20 production; and/or (iii) manufacture the bulk rHuPH20 required by us and our collaborators for use in our proprietary and collaboration products and product candidates. In addition to supply obligations, Avid and Catalent will also provide support for data and information used in the chemistry, manufacturing and controls sections for FDA and other regulatory filings.
We have a commercial manufacturing and supply agreement with Patheon Manufacturing Services, LLC (Patheon) under which Patheon will provide the final fill and finishing steps in the production process of Hylenex recombinant.
9
Sales, Marketing and Distribution
Hylenex Recombinant
Our commercial activities currently focus on Hylenex recombinant. We have a team of sales specialists that provide hospital and surgery center customers with the information about Hylenex recombinant and information needed to obtain formulary approval for, and support utilization of, Hylenex recombinant. Our commercial activities also include marketing and related services and commercial support services such as commercial operations, managed markets and commercial analytics. We also employ third-party vendors, such as advertising agencies, market research firms and suppliers of marketing and other sales support related services to assist with our commercial activities.
We sell Hylenex recombinant in the U.S. to wholesale pharmaceutical distributors, who sell the product to hospitals and other end-user customers. We engage Integrated Commercialization Solutions (ICS), a division of AmerisourceBergen Specialty Group, a subsidiary of AmerisourceBergen, to act as our exclusive distributor for commercial shipment and distribution of Hylenex recombinant to our customers in the United States. In addition to distribution services, ICS provides us with other key services related to logistics, warehousing, returns and inventory management, contract administration and chargebacks processing and accounts receivable management. In addition, we utilize third parties to perform various other services for us relating to regulatory monitoring, including call center management, adverse event reporting, safety database management and other product maintenance services.
Competition
The pharmaceutical industry is characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary therapeutics. We face competition from a number of sources, some of which may target the same indications as our product or product candidates, including large pharmaceutical companies, smaller pharmaceutical companies, biotechnology companies, academic institutions, government agencies and private and public research institutions, many of which have greater financial resources, drug development experience, sales and marketing capabilities, including larger, well established sales forces, manufacturing capabilities, experience in obtaining regulatory approvals for product candidates and other resources than us. We face competition not only in the commercialization of Hylenex but also for the out-licensing of our ENHANZE technology. Our ENHANZE technology may face increasing competition from alternate approaches and/or emerging technologies to deliver medicines SC. In addition, our collaborators face competition in the commercialization of the product candidates for which the collaborators seek marketing approval from the FDA or other regulatory authorities.
Hylenex Recombinant
Hylenex recombinant is currently the only FDA approved recombinant human hyaluronidase on the market. The competitors for Hylenex recombinant include, but are not limited to, Valeant Pharmaceuticals International, Inc.’s product, Vitrase®, an ovine (ram) hyaluronidase, and Amphastar Pharmaceuticals, Inc.’s product, Amphadase®, a bovine (bull) hyaluronidase.
Government Regulations
The FDA and comparable regulatory agencies in foreign countries regulate the manufacture and sale of the pharmaceutical products that we or our partners have developed or that our partners currently are developing. The FDA has established guidelines and safety standards that are applicable to the laboratory and preclinical evaluation and clinical investigation of therapeutic products and stringent regulations that govern the manufacture and sale of these products. The process of obtaining regulatory approval for a new therapeutic product usually requires a significant amount of time and substantial resources.
Regulatory obligations continue post-approval and include the reporting of adverse events when a drug is utilized in the broader patient population. Promotion and marketing of drugs is also strictly regulated, with penalties imposed for violations of FDA regulations, the Lanham Act and other federal and state laws, including the federal anti-kickback statute.
We currently intend to continue to seek, through our collaborators, approval to market products and product candidates in foreign countries, which may have regulatory processes that differ materially from those of the FDA. Our partners may rely upon independent consultants to seek and gain approvals to market our proposed products in foreign countries or may rely on other pharmaceutical or biotechnology companies to license our proposed products. We cannot guarantee that approvals to market any of our partners’ products can be obtained in any country. Approval to market a product in any one foreign country does not necessarily indicate that approval can be obtained in other countries.
From time to time, legislation is drafted and introduced in Congress that could significantly change the statutory provisions governing the approval, manufacturing and marketing of drug products. In addition, FDA regulations and guidance are often revised or reinterpreted by the agency or reviewing courts in ways that may significantly affect our business and development of our partners’ product candidates and any products that we may commercialize. It is impossible to predict
10
whether additional legislative changes will be enacted, or FDA regulations, guidance or interpretations changed, or what the impact of any such changes may be.
Information about our Executive Officers
Information concerning our executive officers, including their names, ages and certain biographical information can be found in Part III, Item 10, Directors, Executive Officers and Corporate Governance. This information is incorporated by reference into Part I of this report.
Human Capital Management
The experience, expertise and dedication of our employees drive the progress and accomplishments of Halozyme.
As of February 17, 2021, we had 136 full-time employees. None of our employees are unionized and we believe our employee relations to be good.
Recognizing the value of our employees and the contributions they make in achieving our business objectives and overall success, we focus on creating and providing an inclusive and safe work environment where employees are respected and rewarded for their contributions, work together as one team, have opportunities to grow and develop their careers, and support the communities in which we work. We also believe this approach to human capital management is essential to attracting and retaining employees in the highly competitive biotechnology and pharmaceutical labor market. To achieve this supportive working environment, our human capital management efforts focus on:
Corporate Values and Ethics:
The foundation of our human capital management strategy is contained in our corporate values statement and our Code of Conduct and Ethics (the “Code of Conduct”), both of which provide uniform guidance to all our employees regarding expectations for proper workplace behavior. Our corporate values emphasize respecting and valuing fellow team members and acting with integrity and honesty to uphold the highest ethical standards. We believe these values provide an environment in which all employees can feel proud and motivated to contribute their valued talents to achieving corporate goals and objectives. Our values also emphasize empowering employees and personal accountability as a means to fulfill our commitments to patients, partners, shareholders and each other.
Our Board of Directors adopted and regularly reviews the Code of Conduct, which applies to all of our employees, officers and directors. Adherence to the Code of Conduct helps ensure that all employees can feel a part of an organization that emphasizes adherence to laws and policies covering the industry in which we work. Our Code of Conduct also emphasizes each employee’s accountability for making decisions and taking actions in a highly ethical manner with a focus on honesty, fairness and integrity and treating all fellow employees in a respectful and inclusive manner. We have established a reporting hotline that enables employees to file anonymous reports of any suspected violations of the Code of Conduct. We believe that providing an ethical environment in which to work is vital to our efforts to attract, retain and develop our employees.
Diversity and Inclusion:
We seek to build and maintain a diverse team of employees that is passionate about and committed to having a positive impact on the lives of patients and their families. We value and celebrate the unique talents, backgrounds and perspectives each employee contributes to achieving our mission and corporate objectives. In support of this philosophy, we adopted the Biotechnology Innovation Organization’s principles on workforce development, diversity and inclusion. Our diverse and inclusive culture is key to attract, develop and retain our talent pool within the globally competitive biotechnology industry. Our dedication to these principles has resulted in a diverse and inclusive employee base consisting of 51% female and 40% non-white/Caucasian employees as of February 17, 2021.
As an equal opportunity employer, we strive to attract and connect with diverse talent who best match our core values and who will be successful and thrive at Halozyme. Our recruiting team partners with hiring managers and with our diverse interview panels to provide support at every stage of the process. We strive to ensure we evaluate a diverse group of candidates for every role with the goal of identifying the best possible candidate – whether internal or external – to fill open roles in the company. We are also committed to providing a positive candidate experience and endeavor to be transparent and respect the time each candidate gives us throughout the process. We evaluate our recruitment and retention efforts based on a variety of metrics, including offer acceptance rate, time-to-hire, turnover and diversity of our employees.
Professional Development for Employees at All Levels:
We are firmly committed to employee development because it is essential to the future growth and overall success of Halozyme. We understand that high performing employees are always searching for ways to broaden and develop their skills. To support our employees, we conduct an individual development plan process to give employees the opportunity and accountability to document their career goals and the actions necessary to achieve those goals. Our senior leader development program is focused on advancing business acumen and leadership skills. Our talent development curriculum for the entire organization is focused on professional, team and leadership development opportunities and grounded in our established leadership attributes which identify the knowledge, skills, abilities and behaviors that contribute to individual and organizational performance. In addition, we provide an online learning platform where employees can access training or
11
programs on a variety of topics whenever they choose. We offer education assistance for college and university courses, training seminars and educational conferences to all employees.
To monitor progress, we review our succession plan for key senior management positions as part of our annual talent review and identify development opportunities to help ensure potential successor readiness.
Employee Engagement:
In order to ensure we are delivering on our human capital initiatives and better understand our employees’ experience, we regularly conduct employee engagement surveys. In 2019, we achieved a 97% response rate with our survey with more than 90% of our employees indicating:
•They are aware of the company mission and vision and have a clear understanding of the goals and objectives of the company;
•Their team is committed to doing high quality work; and
•Their manager treats them with respect and cares about them.
With the goal of continuous performance improvement we are focused on further strengthening cross-functional team work including how team members communicate and hold each other accountable. Examples of specific actions we have taken in response to employee survey feedback include institution of all-employee training on cross-functional teamwork and how to have crucial conversations.
We hold frequent all-employee meetings that serve as a forum to share progress on strategy and corporate goals, celebrate achievements, and share best practices and learnings. In 2020 and continuing in 2021 we have increased the frequency of our all-employee meetings from monthly to every other week while implementing our work-from-home strategy in response to COVID-19, in order to keep employees well-informed and connected and to addresses their questions.
Management tracks and assesses retention and attrition and interviews departing employees in order to identify any addressable trends.
Compensation & Benefits:
Our compensation and benefits programs, with oversight from the Compensation Committee of our Board of Directors, are designed to attract, retain and reward employees through competitive salaries, annual bonus eligibility, stock awards grants to all employees, a 401(k) Plan, healthcare and insurance benefits, health savings and flexible spending accounts, paid time off, family leave, and employee assistance programs. Each year we conduct surveys to benchmark our salaries and benefits and confirm we are satisfied with the competitiveness of our total compensation offering. We also provide a variety of peer-to-peer and corporate recognition programs to celebrate and recognize our employees for their hard work and contributions.
Employee Health and Safety:
We are dedicated to promoting the health and safety of our employees because we believe it fosters employee productivity and job performance. We have developed and implemented annual workplace safety training which is intended to remind our employees of workplace safety procedures that may be useful in the event of emergency situations and to assist our employees in helping to prevent workplace accidents. We have established a Safety Committee which meets on a quarterly basis to review workplace safety and adherence to safety policies. Further, our Code of Conduct emphasizes our commitment to preventing unlawful employment discrimination and workplace harassment including mandatory, on going sexual harassment training and provides a mechanism for reporting any violations of this policy.
Our response to COVID-19:
Because we take the health and safety of our employees, their families and our local communities very seriously, we have implemented the following actions to protect against the transmission of COVID-19 in our office and in the local community, while ensuring that critical work continues:
•Restricted access to our office to only Halozyme personnel necessary to carry out essential activities. All other employees are currently working from home.
•Required self-health check, and on-site temperature screening for all on-site personnel.
•Required virtual, rather than in-person, interactions to continue to meet the needs of our customers, partners and contractors.
•Restricted all air travel.
•Disinfection of all common areas, door handles, restrooms, and kitchens multiple times daily, and regular after-hours disinfection of all non-laboratory areas with state-of-the-art electrostatic sanitization.
•Upgraded all HVAC systems to MERV-13 filtration throughout our campus and introduction of supplementary HEPA filtration in conference rooms and select open office areas.
Corporate Citizenship:
We believe in supporting the community in which we work and provide our employees multiple opportunities to contribute to the community, including providing company-wide community service days/volunteerism supporting:
•STEM education;
12
•Human services (e.g., food drives, home builds, meal services);
•Environmental organizations (e.g., lagoon cleanup events, park restoration);
•Children/Military (e.g., school supply drives, holiday adopt-a-family, playhouse builds, paracord builds);
•Cultural/diversity relations; and
•Patient/healthcare advocacy groups.
13
Item 1A.Risk Factors
Risks Related To Our Business
Business interruptions resulting from the COVID-19 outbreak or similar public health crises could cause a disruption of the development of our collaboration partners’ product candidates and commercialization of approved products, impede our ability to supply bulk rHuPH20 to our partners or procure and sell Hylenex and otherwise adversely impact our business and results of operations.
Public health crises such as pandemics or similar outbreaks could adversely impact our business and results of operations by, among other things, disrupting the development of our collaboration partners’ product candidates and commercialization of our partners’ approved products, disrupting our ability to enter into new ENHANZE collaborations with potential partners in a timely manner, causing disruptions in the operations of our third party contract manufacturing organizations upon whom we rely for the production and supply of our commercial product Hylenex and the bulk rHuPH20 we supply to our partners, and causing other disruptions to our operations. For example, the outbreak of a coronavirus, which causes COVID-19, has rapidly evolved into a global pandemic and has spread to most regions of the world including the city of San Diego, California where our main office is located.
The COVID-19 pandemic is evolving, and to date has led to the implementation of various responses, including government-imposed quarantines, travel restrictions and other public health safety measures. The extent to which COVID-19 and its variants impacts our operations and/or those of our collaboration partners will depend on future developments, which are highly uncertain and unpredictable, including the duration or recurrence of the outbreak, additional or modified government actions, new information that will emerge concerning the severity and impact of COVID-19 and the actions to contain COVID-19 or address its impact in the short and long term, among others.
We have responded to the COVID-19 pandemic by taking a number of actions including closing our offices in San Diego in March 2020, requesting that most of our personnel work remotely and restricting access to our facilities mostly to personnel who perform critical activities that must be completed on-site in accordance with California’s initial statewide shelter-in-place order and ongoing guidances. Increased reliance on personnel working from home may have a negative impact on productivity, or disrupt, delay or otherwise adversely impact business, by, among other things, increasing cyber security risk, impeding access to information that would be helpful to pursue our business objectives or disrupting our communications. We are continuing to monitor the situation and currently do not have an estimate of the timing for all employees returning to the office for work.
The business disruptions associated with a global pandemic could impact the business, product development priorities and operations of our collaboration partners, including potential delays in manufacturing their product candidates or approved products. For example, some of our collaboration partners are conducting or are planning to conduct clinical trials in geographies affected by the COVID-19 pandemic. The progress or completion of these clinical trials could be adversely impacted by the pandemic. Additionally, interruption or delays in the operations of the FDA, the EMA and other similar foreign regulatory agencies, or changes in regulatory priorities to focus on the COVID-19 pandemic, may affect required regulatory review, inspection, clearance and approval timelines. Disruptions such as these could result in delays in the development programs of our collaboration products or impede the commercial efforts for approved products, resulting in potential reductions or delays in our revenues from collaborator royalty or milestone payments. We do not know the extent to which our collaboration partners’ development programs or product commercialization efforts will be impacted or delayed.
We rely on third party manufacturers to manufacture the bulk rHuPH20 that we supply to our collaboration partners for their commercial products and product candidates, as well as our commercial product Hylenex. If any such third party manufacturer is adversely impacted by the COVID-19 pandemic and related consequences, including staffing shortages, production slowdowns and disruptions in delivery systems, availability of raw materials, reagents or components or if they divert resources or manufacturing capacity to accommodate the development of coronavirus treatments or vaccines, our supply chain may be disrupted, limiting our ability to sell Hylenex or supply bulk rHuPH20 to our collaboration partners. Any such disruptions could result in reductions or delays in our revenues.
The effects of COVID-19 could worsen in countries that are already afflicted with the coronavirus which could further adversely impact our ability to conduct our business and could have a material adverse impact on our operations, financial condition and results. We do not yet know the full extent of the impact that COVID-19 may or will have on our business.
In addition, the trading prices for our common shares and other biopharmaceutical companies have been highly volatile as a result of market and investor reactions to the COVID-19 pandemic and its potential consequences. As a result, access to sources of financing, should those be needed, may be more difficult and/or expensive. In addition, a recession, depression or
14
other sustained adverse market event resulting from the spread of the coronavirus could materially and adversely affect our business and the value of our common shares.
We have generated only limited revenues to date and we have a history of net losses and negative cash flows.
Relative to expenses incurred in our operations, we have generated only limited revenues from product sales, royalties, licensing fees, milestone payments, bulk rHuPH20 supply payments and research reimbursements to date. Through December 31, 2020, we have incurred aggregate net losses of $474.6 million. Although we expect to maintain sustainable profitability, unexpected declines in revenues and increases in expenses could inhibit our ability to sustain profitability.
If partners’ product candidates do not receive and maintain regulatory approvals, or if approvals are not obtained in a timely manner, such failure or delay would substantially impair our ability to generate revenues.
Approval from the FDA or equivalent health authorities is necessary to manufacture and market pharmaceutical products in the U.S. and the other countries in which we anticipate doing business have similar requirements. The process for obtaining FDA and other regulatory approvals is extensive, time-consuming, risky and costly, and there is no guarantee that the FDA or other regulatory bodies will approve any applications that may be filed with respect to any of our partners’ product candidates, or that the timing of any such approval will be appropriate for the desired product launch schedule for a product candidate. We and our collaborators attempt to provide guidance as to the timing for the filing and acceptance of such regulatory approvals, but such filings and approvals may not occur when we or our collaborators expect, or at all. The FDA or other foreign regulatory agency may refuse or delay approval of our partners’ product candidates for failure to collect sufficient clinical or animal safety data and require our collaborators to conduct additional clinical or animal safety studies which may cause lengthy delays and increased costs to our partners’ development programs. Any such issues associated with rHuPH20 could have an adverse impact on future development of our partners’ products which include rHuPH20, future sales of Hylenex recombinant, or our ability to maintain our existing collaborations or enter into new collaborations.
We and our collaborators may not be successful in obtaining approvals for any additional potential products in a timely manner, or at all. Refer to the risk factor titled “Our collaboration product candidates may not receive regulatory approvals or their development may be delayed for a variety of reasons, including delayed or unsuccessful clinical trials, regulatory requirements or safety concerns” for additional information relating to the approval of product candidates.
Additionally, even with respect to products which have been approved for commercialization, in order to continue to manufacture and market pharmaceutical products, we or our collaborators must maintain our regulatory approvals. If we or any of our collaborators are unsuccessful in maintaining our regulatory approvals, our ability to generate revenues would be adversely affected.
Use of Hylenex and the products and product candidates of our partners’ could be associated with side effects or adverse events.
As with most pharmaceutical products, use of Hylenex and the products and product candidates of our collaborators could be associated with side effects or adverse events which can vary in severity (from minor reactions to death) and frequency (infrequent or prevalent). Side effects or adverse events associated with the use of Hylenex and the products or product candidates of our collaborators may be observed at any time, including in clinical trials or when a product is commercialized, and any such side effects or adverse events may negatively affect our or our collaborators’ ability to obtain or maintain regulatory approval or market such products and product candidates. Side effects such as toxicity or other safety issues associated with the use of Hylenex and the products and product candidates of our collaborators could require us or our collaborators to perform additional studies or halt development or commercialization of these products and product candidates or expose us to product liability lawsuits which will harm our business. For example, several years ago we experienced a clinical hold on patient enrollment and dosing in our phase 2 study of PEGPH20 in patients with PDA (a discontinued program), which was not resolved until we implemented steps to address an observed possible difference in TE event rates between the arms of the study. We or our collaborators may be required by regulatory agencies to conduct additional animal or human studies regarding the safety and efficacy of our pharmaceutical products or product candidates which we have not planned or anticipated. Furthermore, there can be no assurance that we or our collaborators will resolve any issues related to any product or product candidate side effects or adverse events to the satisfaction of the FDA or any regulatory agency in a timely manner or ever, which could harm our business, prospects and financial condition.
If our contract manufacturers or vendors are unable to manufacture and supply to us bulk rHuPH20 or other raw materials, reagents or components in the quantity and quality required by us or our collaborators for use in the production of Hylenex or our partners’ products and product candidates, our Hylenex Commercialization efforts or our partners’ product development and commercialization efforts could be delayed or stopped and our business and results of operations and our collaborations could be harmed.
We have existing supply agreements with contract manufacturing organizations Avid Bioservices, Inc. (Avid) and Catalent Indiana LLC (Catalent) to produce bulk rHuPH20. These manufacturers each produce bulk rHuPH20 under cGMP for
15
use in Hylenex recombinant, and for use in collaboration products and product candidates. We rely on their ability to successfully manufacture bulk rHuPH20 according to product specifications. In addition to supply obligations, our contract manufacturers also provide support for the chemistry, manufacturing and controls sections for FDA and other regulatory filings. We also rely on vendors to supply us with raw materials to produce reagents and other materials for bioanalytical assays used to support our partners’ clinical trials. We also have a commercial manufacturing and supply agreement with Patheon under which Patheon provides the final fill and finishing steps in the production process of Hylenex recombinant. If any of our contract manufacturers or vendors: (i) is unable to retain its status as an FDA approved manufacturing facility; (ii) is unable to otherwise successfully scale up bulk rHuPH20 production to meet corporate or regulatory authority quality standards; (iii) is unable to procure raw materials, reagents or components necessary to produce bulk rHuPH20, Hylenex recombinant or our bioanalytical assays or (iv) fails to manufacture and supply bulk rHuPH20 in the quantity and quality required by us or our collaborators for use in Hylenex and collaboration products and product candidates for any other reason, our business will be adversely affected. In addition, a significant change in such parties’ or other third party manufacturers’ business or financial condition could adversely affect their abilities to fulfill their contractual obligations to us. We have not established, and may not be able to establish, favorable arrangements with additional bulk rHuPH20 manufacturers and suppliers of the ingredients necessary to manufacture bulk rHuPH20 should the existing manufacturers and suppliers become unavailable or in the event that our existing manufacturers and suppliers are unable to adequately perform their responsibilities. We have attempted to mitigate the impact of a potential supply interruption through the establishment of excess bulk rHuPH20 inventory where possible, but there can be no assurances that this safety stock will be maintained or that it will be sufficient to address any delays, interruptions or other problems experienced by any of our contract manufacturers. Any delays, interruptions or other problems regarding the ability of any of our contract manufacturers to supply bulk rHuPH20 or the ability of other third party manufacturers, to supply other raw materials or ingredients necessary to produce our products on a timely basis could: (i) cause the delay of our partners’ clinical trials or otherwise delay or prevent the regulatory approval of our partners’ product candidates; (ii) delay or prevent the effective commercialization of Hylenex or collaboration products and product candidates; and/or (iii) cause us to breach contractual obligations to deliver bulk rHuPH20 to our collaborators. Such delays would likely damage our relationship with our collaborators, and they would have a material adverse effect on royalties and thus our business and financial condition. Additionally, we rely on third parties to manufacture, prepare, fill, finish, package, store and ship our product and partners’ product candidates on our behalf. If the third parties we identify fail to perform their obligations, the progress of partners’ clinical trials could be delayed or even suspended and the commercialization of our product and partner products could be delayed or prevented.
If we or any party to a key collaboration agreement fail to perform material obligations under such agreement, or if a key collaboration agreement, is terminated for any reason, our business could significantly suffer.
We have entered into multiple collaboration agreements under which we may receive significant future payments in the form of milestone payments, target designation fees, maintenance fees and royalties. We are heavily dependent on our collaborators to develop and commercialize product candidates subject to our collaborations in order for us to realize any financial benefits from these collaborations. Our collaborators may not devote the attention and resources to such efforts that we would ourselves, change their clinical development plans, promotional efforts or simultaneously develop and commercialize products in competition to those products we have licensed to them. Any of these actions could not be visible to us immediately and could negatively impact our ability to forecast and our ability to achieve the benefits and revenue we receive from such collaboration. In addition, in the event that a party fails to perform under a key collaboration agreement, or if a key collaboration agreement is terminated, the reduction in anticipated revenues could negatively impact our operations. In addition, the termination of a key collaboration agreement by one or more of our collaborators could have a material adverse impact our ability to enter into additional collaboration agreements with new collaborators on favorable terms, if at all. In certain circumstances, the termination of a key collaboration agreement would require us to revise our corporate strategy going forward and reevaluate the applications and value of our technology.
Hylenex and our partners’ products and product candidates rely on the rHuPH20 enzyme, and any adverse development regarding rHuPH20 could substantially impact multiple areas of our business, including current and potential collaborations, as well as any proprietary programs.
rHuPH20 is a key technological component of Hylenex and our ENHANZE technology and most of our collaboration products and product candidates, including the current and future products and product candidates under our ENHANZE collaborations. If there is an adverse development for rHuPH20 (e.g., an adverse regulatory determination relating to rHuPH20, if we are unable to obtain sufficient quantities of rHuPH20, if we are unable to obtain or maintain material proprietary rights to rHuPH20 or if we discover negative characteristics of rHuPH20), multiple areas of our business, including current and potential collaborations, as well as proprietary programs would be substantially impacted. For example, elevated anti-rHuPH20 antibody titers were detected in the registration trial for Baxalta’s HYQVIA product as well as in a former collaborator’s product in a Phase 2 clinical trial with rHuPH20, but have not been associated, in either case, with any adverse events. We monitor for antibodies to rHuPH20 in our collaboration and proprietary programs, and although we do not believe at this time that the incidence of non-neutralizing anti-rHuPH20 antibodies in either the HYQVIA program or the former collaborator’s program
16
will have a significant impact on our proprietary product and our partners’ product and product candidates, there can be no assurance that there will not be other such occurrences in the foregoing programs or that concerns regarding these antibodies will not also be raised by the FDA or other health authorities in the future, which could result in delays or discontinuations of our Hylenex commercialization activities, the development or commercialization activities of our partners, or deter our entry into additional collaborations with third parties.
Our business strategy and our strategic focus is currently limited to only a few fields or applications of our technology which may increase the risk for potential negative impact from adverse developments. Future expansion of our strategic focus to additional applications of our technology may require the use of additional resources, result in increased expense and ultimately may not be successful.
We routinely evaluate our business strategy, and may modify this strategy in the future in light of our assessment of unmet medical needs, growth potential, resource requirements, regulatory issues, competition, risks and other factors. As a result of these strategic evaluations, we may focus our resources and efforts on one or a few programs or fields and may suspend or reduce our efforts on other programs and fields. For example, in the fourth quarter of 2019, we decided to focus our resources on our ENHANZE technology and our commercial product, Hylenex. By focusing on these areas, we increase the potential impact on us if one of those partner programs does not successfully complete clinical trials, achieve commercial acceptance or meet expectations regarding sales and revenue. We may also expand our strategic focus by seeking new therapeutics applications of our technology which may require the use of additional resources, increased expense and would require the attention of senior management. There can be no assurance that any such investment of resources would ultimately result in additional approved collaboration products or commercial success of new therapeutic applications of our technology.
Our collaboration product candidates may not receive regulatory approvals or their development may be delayed for a variety of reasons, including delayed or unsuccessful clinical trials, regulatory requirements or safety concerns.
Clinical testing of pharmaceutical products is a long, expensive and uncertain process, and the failure or delay of a clinical trial can occur at any stage, including the patient enrollment stage. Even if initial results of preclinical and nonclinical studies or clinical trial results are promising, our collaborators may obtain different results in subsequent trials or studies that fail to show the desired levels of dose safety and efficacy, or our collaborators may not, obtain applicable regulatory approval for a variety of other reasons. Preclinical, nonclinical, and clinical trials for collaboration product candidates could be unsuccessful, which would delay or preclude regulatory approval and commercialization of the product candidates. In the U.S. and other jurisdictions, regulatory approval can be delayed, limited or not granted for many reasons, including, among others:
•during the course of clinical studies, the final data may differ from initial reported data, and clinical results may not meet prescribed endpoints for the studies or otherwise provide sufficient data to support the efficacy of our collaborators’ product candidates;
•clinical and nonclinical test results may reveal inferior pharmacokinetics, side effects, adverse events or unexpected safety issues associated with the use of our collaborators’ product candidates;
•regulatory review may not find that the data from preclinical testing and clinical trials justifies approval;
•regulatory authorities may require that our partners change their studies or conduct additional studies which may significantly delay or make continued pursuit of approval commercially unattractive;
•a regulatory agency may reject partner trial data or disagree with their interpretations of either clinical trial data or applicable regulations;
•a regulatory agency may require additional safety monitoring and reporting through Risk Evaluation and Mitigation Strategies or conditions to assure safe use programs;
•a partner may decide to not pursue regulatory approval for such a product;
•a regulatory agency may not approve our manufacturing processes or facilities, or the processes or facilities of our collaborators, our contract manufacturers or our raw material suppliers;
•a regulatory agency may identify problems or other deficiencies in our existing manufacturing processes or facilities, or the existing processes or facilities of our collaborators, our contract manufacturers or our raw material suppliers;
•a regulatory agency may change its formal or informal approval requirements and policies, act contrary to previous guidance, adopt new regulations or raise new issues or concerns late in the approval process; or
•a partner product candidate may be approved only for indications that are narrow or under conditions that place the product at a competitive disadvantage, which may limit the sales and marketing activities for such product candidate or otherwise adversely impact the commercial potential of a product.
If a collaboration product candidate is not approved in a timely fashion or obtained on commercially viable terms, or if development of any product candidate is terminated due to difficulties or delays encountered in the regulatory approval process, it could have a material adverse impact on our business, and we would become more dependent on the development of other collaboration product candidates and/or our ability to successfully acquire other technologies. There can be no assurances that any collaboration product candidate will receive regulatory approval in a timely manner, or at all. There can be no assurance that partners will be able to gain clarity as to the FDA’s requirements or that the requirements may be satisfied in a
17
commercially feasible way, in which case our ability to enter into collaborations with third parties or explore other strategic alternatives to exploit an opportunity will be limited or may not be possible.
We anticipate that certain collaboration products will be marketed, and perhaps manufactured, in foreign countries. The process of obtaining regulatory approvals in foreign countries is subject to delay and failure for the reasons set forth above, as well as for reasons that vary from jurisdiction to jurisdiction. The approval process varies among countries and jurisdictions and can involve additional testing. The time required to obtain approval may differ from that required to obtain FDA approval. Foreign regulatory agencies may not provide approvals on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or jurisdictions or by the FDA.
Our third party collaborators are responsible for providing certain proprietary materials that are essential components of our collaboration products and product candidates, and any failure to supply these materials could delay the development and commercialization efforts for these collaboration products and product candidates and/or damage our collaborations.
Our development and commercialization collaborators are responsible for providing certain proprietary materials that are essential components of our collaboration products and product candidates. For example, Roche is responsible for producing the Herceptin and MabThera required for its subcutaneous products and Baxalta is responsible for producing the GAMMAGARD LIQUID for its product HYQVIA. If a collaborator, or any applicable third party service provider of a collaborator, encounters difficulties in the manufacture, storage, delivery, fill, finish or packaging of the collaboration product or product candidate or component of such product or product candidate, such difficulties could (i) cause the delay of clinical trials or otherwise delay or prevent the regulatory approval of collaboration product candidates; and/or (ii) delay or prevent the effective commercialization of collaboration products. Such delays could have a material adverse effect on our business and financial condition.
If we or our collaborators fail to comply with regulatory requirements applicable to promotion, sale and manufacturing of approved products, regulatory agencies may take action against us or them, which could significantly harm our business.
Any approved products, along with the manufacturing processes, post-approval clinical data, labeling, advertising and promotional activities for these products, are subject to continual requirements and review by the FDA, state and foreign regulatory bodies. Regulatory authorities subject a marketed product, its manufacturer and the manufacturing facilities to continual review and periodic inspections. We, our collaborators and our respective contractors, suppliers and vendors, will be subject to ongoing regulatory requirements, including complying with regulations and laws regarding advertising, promotion and sales of drug products, required submissions of safety and other post-market information and reports, registration requirements, cGMP regulations (including requirements relating to quality control and quality assurance, as well as the corresponding maintenance of records and documentation), and the requirements regarding the distribution of samples to physicians and recordkeeping requirements. Regulatory agencies may change existing requirements or adopt new requirements or policies. We, our collaborators and our respective contractors, suppliers and vendors, may be slow to adapt or may not be able to adapt to these changes or new requirements.
In particular, regulatory requirements applicable to pharmaceutical products make the substitution of suppliers and manufacturers costly and time consuming. We have minimal internal manufacturing capabilities and are, and expect to be in the future, entirely dependent on contract manufacturers and suppliers for the manufacture of our products and for their active and other ingredients. The disqualification of these manufacturers and suppliers through their failure to comply with regulatory requirements could negatively impact our business because the delays and costs in obtaining and qualifying alternate suppliers (if such alternative suppliers are available, which we cannot assure) could delay our partners’ clinical trials or otherwise inhibit our or partners’ ability to bring approved products to market, which would have a material adverse effect on our business and financial condition. Likewise, if we, our collaborators and our respective contractors, suppliers and vendors involved in sales and promotion of our products do not comply with applicable laws and regulations, for example off-label or false or misleading promotion, this could materially harm our business and financial condition.
Failure to comply with regulatory requirements may result in any of the following:
•restrictions on our or our partners’ products or manufacturing processes;
•warning letters;
•withdrawal of our or our partners’ products from the market;
•voluntary or mandatory recall;
•fines;
•suspension or withdrawal of regulatory approvals;
•suspension or termination of any of our partners’ ongoing clinical trials;
•refusal to permit the import or export of our or our partners’ products;
•refusal to approve pending applications or supplements to approved applications that we submit;
•product seizure;
•injunctions; or
18
•imposition of civil or criminal penalties.
We may need to raise additional capital in the future and there can be no assurance that we will be able to obtain such funds.
We may need to raise additional capital in the future to fund our operations and for general corporate purposes if revenues do not occur as expected. Our current cash reserves and expected revenues may not be sufficient for us to fund general operations and conduct our business at the level desired. In addition, if we engage in acquisitions of companies, products or technologies in order to execute our business strategy, we may need to raise additional capital. We may raise additional capital in the future through one or more financing vehicles that may be available to us including (i) new collaborative agreements; (ii) expansions or revisions to existing collaborative relationships; (iii) private financings; (iv) other equity or debt financings; (v) monetizing assets; and/or (vi) the public offering of securities.
If we are required to raise additional capital in the future, it may not be available on favorable financing terms within the time required, or at all. If additional capital is not available on favorable terms when needed, we will be required to raise capital on adverse terms or significantly reduce operating expenses through the restructuring of our operations or deferral of strategic business initiatives. If we raise additional capital through a public offering of securities or equity, a substantial number of additional shares may be issued, which may negatively affect our stock price and these additional shares will dilute the ownership interest of our current investors.
We currently have significant debt and may incur additional debt. Failure by us to fulfill our obligations under the applicable debt agreements may cause the repayment obligations to accelerate.
The aggregate amount of our consolidated indebtedness, net of debt discount, as of December 31, 2020 was $397.2 million, which includes $460.0 million in aggregate principal amount of 1.25% Convertible Senior Notes due 2024 (Convertible Notes) net of unamortized debt discount of $62.8 million. We also may incur additional indebtedness in the future.
Our indebtedness may:
•make it difficult for us to satisfy our financial obligations, including making scheduled principal and interest payments on our indebtedness;
•limit our ability to borrow additional funds for working capital, capital expenditures, acquisitions or other general corporate purposes;
•limit our ability to use our cash flow or obtain additional financing for future working capital, capital expenditures, acquisitions, share repurchases or other general business purposes;
•require us to use a portion of our cash flow from operations to make debt service payments;
•limit our flexibility to plan for, or react to, changes in our business and industry;
•place us at a competitive disadvantage compared to our less leveraged competitors; and
•increase our vulnerability to the impact of adverse economic and industry conditions.
Our ability to make payments on our existing or any future debt will depend on our future operating performance and ability to generate cash and may also depend on our ability to obtain additional debt or equity financing. It will also depend on financial, business or other factors affecting our operations, many of which are beyond our control. We will need to use cash to pay principal and interest on our debt, thereby reducing the funds available to fund operations, strategic initiatives and working capital requirements. If we are unable to generate sufficient cash to service our debt obligations, an event of default may occur under any of our debt instruments which could result in an acceleration of such debt upon which we may be required to repay all the amounts outstanding under some or all of our debt instruments. Such an acceleration of our debt obligations could harm our financial condition. From time to time, we may seek to retire or repurchase our outstanding debt through cash purchases and/or exchanges for equity or debt, in open-market purchases, privately negotiated transactions or otherwise. Any such repurchases or exchanges would be on such terms and at such prices as we determine, and will depend on current market conditions, our liquidity needs, any restrictions in our contracts and other factors. The amounts involved in such transactions could be material.
The conditional conversion feature of the Convertible Notes, if triggered, may adversely affect our financial condition and operating results.
In the event the conditional conversion feature of the Convertible Notes is triggered, holders of the Convertible Notes will be entitled to convert the notes at any time during specified periods at their option. If one or more holders elect to convert their notes, we would be required to settle a portion or all of our conversion obligation in cash, which could adversely affect our liquidity. Even if holders of the Convertible Notes do not elect to convert their notes, we are required under applicable accounting rules to reclassify all or a portion of the outstanding principal of the notes as a current rather than long-term liability when the conditional conversion feature is triggered, which results in a material reduction of our net working capital. For example, as of December 31, 2020, the conditional conversion feature was triggered and our notes are classified as a current liability.
19
Conversion of our Convertible Notes may dilute the ownership interest of existing stockholders or may otherwise depress the price of our common stock.
The conversion of some or all of our Convertible Notes, to the extent we deliver shares upon conversion, will dilute the ownership interests of existing stockholders. Any sales in the public market of the Convertible Notes or our common stock issuable upon conversion of the Convertible Notes could adversely affect prevailing market prices of our common stock. In addition, the existence of the Convertible Notes may encourage short selling by market participants because the conversion of the Convertible Notes could be used to satisfy short positions, or anticipated conversion of the Convertible Notes into shares of our common stock could depress the price of our common stock.
The accounting method for the Convertible Notes could have a material effect on our reported financial results.
Pursuant to Financial Accounting Standards Board Accounting Standards Codification Subtopic 470-20, Debt with Conversion and Other Options (“ASC 470-20”), an entity must separately account for the liability and equity components of convertible debt instruments whose conversion may be settled entirely or partially in cash (such as our Convertible Notes) in a manner that reflects the issuer’s economic interest cost for non-convertible debt. The liability component of our Convertible Notes was initially valued at the fair value of a similar debt instrument that does not have an associated equity component and was reflected as a liability in our consolidated balance sheet. The equity component of the Convertible Notes was included in the additional paid-in capital section of our stockholders’ equity on our consolidated balance sheet, and the value of the equity component was treated as original issue discount for purposes of accounting for the debt component. This original issue discount will be amortized to non-cash interest expense over the term of the notes, and we will record a greater amount of non-cash interest expense in current periods as a result of this amortization. Accordingly, we will report lower net income in our financial results because ASC 470-20 will require the interest expense associated with the notes to include both the current period’s amortization of the debt discount and the notes’ coupon interest, which could adversely affect our reported or future financial results, the trading price of our common stock and the trading price of the Convertible Notes.
If collaboration product candidates are approved for marketing but do not gain market acceptance resulting in commercial performance below that which was expected or projected, our business may suffer and we may not be able to fund future operations.
Assuming that existing or future collaboration product candidates obtain the necessary regulatory approvals for commercial sale, a number of factors may affect the market acceptance of these newly-approved products, including, among others:
•the degree to which the use of these products is restricted by the approved product label;
•the price of these products relative to other therapies for the same or similar treatments;
•the extent to which reimbursement for these products and related treatments will be available from third party payors including government insurance programs and private insurers;
•the introduction of generic or biosimilar competitors to these products;
•the perception by patients, physicians and other members of the health care community of the effectiveness and safety of these products for their prescribed treatments relative to other therapies for the same or similar treatments;
•the ability and willingness of our collaborators to fund sales and marketing efforts; and
•the effectiveness of the sales and marketing efforts of our collaborators.
If these collaboration products do not gain market acceptance resulting in commercial performance below that which was expected or projected, the royalties we expect to receive from these products will be diminished which could harm our ability to fund future operations, including conduct acquisitions, execute our planned share repurchases, or affect our ability to use funds for other general corporate purposes and cause our business to suffer.
In addition, our partners’ product candidates will be restricted to the labels approved by FDA and applicable regulatory bodies, and these restrictions may limit the marketing and promotion of the ultimate products. If the approved labels are restrictive, the sales and marketing efforts for these collaboration products may be negatively affected.
Our ability to license our ENHANZE technology to our collaboration partners depends on the validity of our patents and other proprietary rights.
Patents and other proprietary rights are essential to our business. Our success will depend in part on our ability to obtain and maintain patent protection for our inventions, to preserve our trade secrets and to operate without infringing the proprietary rights of third parties. We have multiple patents and patent applications throughout the world pertaining to our recombinant human hyaluronidase and methods of use and manufacture, including an issued U.S. patent which expires in 2027 and an issued European patent which expires in 2024, which we believe cover the products and product candidates under our existing collaborations, and Hylenex. Although we believe our patent filings represent a barrier to entry for potential competitors looking to utilize these hyaluronidases, upon expiration of our patents other pharmaceutical companies may (if they do not
20
infringe our other patents) seek to compete with us by developing, manufacturing and selling biosimilars to the active drug ingredient in our ENHANZE technology used by our collaboration partners in combination with their products. Any such loss of patent protection or proprietary rights could lead to a reduction or loss of revenues, incentivize one or more of our key ENHANZE collaboration partners to terminate their relationship with us and impact our ability to enter into new collaboration and license agreements.
Developing and marketing pharmaceutical products for human use involves significant product liability risks for which we currently have limited insurance coverage.
The testing, marketing and sale of pharmaceutical products involves the risk of product liability claims by consumers and other third parties. Although we maintain product liability insurance coverage, product liability claims can be high in the pharmaceutical industry, and our insurance may not sufficiently cover our actual liabilities. If product liability claims were to be made against us, it is possible that the liabilities may exceed the limits of our insurance policy, or our insurance carriers may deny, or attempt to deny, coverage in certain instances. If a lawsuit against us is successful, then the insurance coverage may not be sufficient and could materially and adversely affect our business and financial condition. Furthermore, various distributors of pharmaceutical products require minimum product liability insurance coverage before purchase or acceptance of products for distribution. Failure to satisfy these insurance requirements could impede our ability to achieve broad distribution of our proposed products, and higher insurance requirements could impose additional costs on us. In addition, since many of our collaboration product candidates include the pharmaceutical products of a third party, we run the risk that problems with the third party pharmaceutical product will give rise to liability claims against us.
If our collaborators do not achieve projected development, clinical, or regulatory goals in the timeframes publicly announced or otherwise expected, the commercialization of our collaboration products may be delayed and, as a result, our stock price may decline, and we may face lawsuits relating to such declines.
From time to time, our collaborators may publicly articulate the estimated timing for the accomplishment of certain scientific, clinical, regulatory and other product development goals. The accomplishment of any goal is typically based on numerous assumptions, and the achievement of a particular goal may be delayed for any number of reasons both within and outside of our and our collaborators’ control. If scientific, regulatory, strategic or other factors cause a collaboration partner to not meet a goal, regardless of whether that goal has been publicly articulated or not, our stock price may decline rapidly. Stock price declines may also trigger direct or derivative shareholder lawsuits. As with any litigation proceeding, the eventual outcome of any legal action is difficult to predict. If any such lawsuits occur, we will incur expenses in connection with the defense of these lawsuits, and we may have to pay substantial damages or settlement costs in connection with any resolution thereof. Although we have insurance coverage against which we may claim recovery against some of these expenses and costs, the amount of coverage may not be adequate to cover the full amount or certain expenses and costs may be outside the scope of the policies we maintain. In the event of an adverse outcome or outcomes, our business could be materially harmed from depletion of cash resources, negative impact on our reputation, or restrictions or changes to our governance or other processes that may result from any final disposition of the lawsuit. Moreover, responding to and defending pending litigation significantly diverts management’s attention from our operations.
In addition, the consistent failure to meet publicly announced milestones may erode the credibility of our management team with respect to future milestone estimates.
Future acquisitions could disrupt our business and harm our financial condition.
In order to augment our product pipeline or otherwise strengthen our business, we may decide to acquire additional businesses, products and technologies. As we have limited experience in evaluating and completing acquisitions, our ability as an organization to make such acquisitions is unproven. Acquisitions could require significant capital infusions and could involve many risks, including, but not limited to, the following:
•we may have to issue additional convertible debt or equity securities to complete an acquisition, which would dilute our stockholders and could adversely affect the market price of our common stock;
•an acquisition may negatively impact our results of operations because it may require us to amortize or write down amounts related to goodwill and other intangible assets, or incur or assume substantial debt or liabilities, or it may cause adverse tax consequences, substantial depreciation or deferred compensation charges;
•we may encounter difficulties in assimilating and integrating the business, products, technologies, personnel or operations of companies that we acquire;
•certain acquisitions may impact our relationship with existing or potential collaborators who are competitive with the acquired business, products or technologies;
•acquisitions may require significant capital infusions and the acquired businesses, products or technologies may not generate sufficient value to justify acquisition costs;
•we may take on liabilities from the acquired company such as debt, legal liabilities or business risk which could be significant;
21
•an acquisition may disrupt our ongoing business, divert resources, increase our expenses and distract our management;
•acquisitions may involve the entry into a geographic or business market in which we have little or no prior experience; and
•key personnel of an acquired company may decide not to work for us.
If any of these risks occurred, it could adversely affect our business, financial condition and operating results. There is no assurance that we will be able to identify or consummate any future acquisitions on acceptable terms, or at all. If we do pursue any acquisitions, it is possible that we may not realize the anticipated benefits from such acquisitions or that the market will not view such acquisitions positively.
Risks Related To Ownership of Our Common Stock
Our stock price is subject to significant volatility.
We participate in a highly dynamic industry which often results in significant volatility in the market price of common stock irrespective of company performance. The high and low sales prices of our common stock during the twelve months ended December 31, 2020 were $44.53 and $12.71, respectively. We expect our stock price to continue to be subject to significant volatility and, in addition to the other risks and uncertainties described elsewhere in this Annual Report on Form 10-K and all other risks and uncertainties that are either not known to us at this time or which we deem to be immaterial, any of the following factors may lead to a significant drop in our stock price:
•the presence of competitive products to those being developed by our partners;
•failure (actual or perceived) of our collaborators to devote attention or resources to the development or commercialization of products or product candidates licensed to such collaborator;
•a dispute regarding our failure, or the failure of one of our third party collaborators, to comply with the terms of a collaboration agreement;
•the termination, for any reason, of any of our collaboration agreements;
•the sale of common stock by any significant stockholder, including, but not limited to, direct or indirect sales by members of management or our Board of Directors;
•the resignation, or other departure, of members of management or our Board of Directors;
•general negative conditions in the healthcare industry;
•pandemics or other global crises;
•general negative conditions in the financial markets;
•the cost associated with obtaining regulatory approval for any of our collaboration product candidates;
•the failure, for any reason, to secure or defend our intellectual property position;
•the failure or delay of applicable regulatory bodies to approve our partners’ product candidates;
•identification of safety or tolerability issues;
•failure of our partners’ clinical trials to meet efficacy endpoints;
•suspensions or delays in the conduct of our partners’ clinical trials or securing of regulatory approvals;
•adverse regulatory action with respect to our and our collaborators’ products and product candidates such as loss of regulatory approval to commercialize such products, clinical holds, imposition of onerous requirements for approval or product recalls;
•our failure, or the failure of our third party collaborators, to successfully commercialize products approved by applicable regulatory bodies such as the FDA;
•our failure, or the failure of our third party collaborators, to generate product revenues anticipated by investors;
•disruptions in our clinical or commercial supply chains, including disruptions caused by problems with a bulk rHuPH20 contract manufacturer or a fill and finish manufacturer for any product or product collaboration candidate;
•the sale of additional debt and/or equity securities by us;
•our failure to obtain financing on acceptable terms or at all;
•a restructuring of our operations;
•an inability to execute our share repurchase program in the time and manner we expect due to market, business, legal or other considerations; or
•a conversion of the Convertible Notes into shares of our common stock.
Future transactions where we raise capital may negatively affect our stock price.
We are currently a “Well-Known Seasoned Issuer” and may file automatic shelf registration statements at any time with the SEC. Sales of substantial amounts of shares of our common stock or other securities under our current or future shelf registration statements could lower the market price of our common stock and impair our ability to raise capital through the sale of equity securities.
22
Anti-takeover provisions in our charter documents, the Indenture and Delaware law may make an acquisition of us more difficult.
Anti-takeover provisions in our charter documents, the Indenture and Delaware law may make an acquisition of us more difficult. First, our Board of Directors is classified into three classes of directors. Under Delaware law, directors of a corporation with a classified board may be removed only for cause unless the corporation’s certificate of incorporation provides otherwise. Our amended and restated certificate of incorporation, as amended, does not provide otherwise. In addition, our bylaws limit who may call special meetings of stockholders, permitting only stockholders holding at least 50% of our outstanding shares to call a special meeting of stockholders. Our amended and restated certificate of incorporation does not include a provision for cumulative voting for directors. Under cumulative voting, a minority stockholder holding a sufficient percentage of a class of shares may be able to ensure the election of one or more directors. Finally, our bylaws establish procedures, including advance notice procedures, with regard to the nomination of candidates for election as directors and stockholder proposals.
These provisions in our charter documents may discourage potential takeover attempts, discourage bids for our common stock at a premium over market price or adversely affect the market price of, and the voting and other rights of the holders of, our common stock. These provisions could also discourage proxy contests and make it more difficult for stockholders to elect directors other than the candidates nominated by our board of directors.
Further, in connection with our recent Convertible Notes issuance, we entered into an indenture, dated as of November 18, 2019, (“Indenture”) with The Bank of New York Mellon Trust Company, N.A., as trustee. Certain provisions in the Indenture could make it more difficult or more expensive for a third party to acquire us. For example, if a takeover would constitute a fundamental change, holders of the Convertible Notes will have the right to require us to repurchase their Convertible Notes in cash. In addition, if a takeover constitutes a make-whole fundamental change, we may be required to increase the conversion rate for holders who convert their Convertible Notes in connection with such takeover. In either case, and in other cases, our obligations under the Convertible Notes and the Indenture could increase the cost of acquiring us or otherwise discourage a third party from acquiring us or removing incumbent management
In addition, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which may prohibit large stockholders from consummating a merger with, or acquisition of, us.
These provisions may deter an acquisition of us that might otherwise be attractive to stockholders.
Risks Related to Our Industry
Our partners’ products must receive regulatory approval before they can be sold, and compliance with the extensive government regulations is expensive and time consuming and may result in the delay or cancellation of collaboration product sales, introductions or modifications.
Extensive industry regulation has had, and will continue to have, a significant impact on our business. All pharmaceutical companies, including ours, are subject to extensive, complex, costly and evolving regulation by the health regulatory agencies including the FDA (and with respect to controlled drug substances, the U.S. Drug Enforcement Administration (DEA)) and equivalent foreign regulatory agencies and state and local/regional government agencies. The Federal Food, Drug and Cosmetic Act, the Controlled Substances Act and other domestic and foreign statutes and regulations govern or influence the testing, manufacturing, packaging, labeling, storing, recordkeeping, safety, approval, advertising, promotion, sale and distribution of our product and our partners’ products and product candidates. We are dependent on receiving FDA and other governmental approvals, including regulatory approvals in jurisdictions outside the United States, prior to manufacturing, marketing and shipping our products. Consequently, there is always a risk that the FDA or other applicable governmental authorities, including those outside the United States, will not approve our partners’ products or may impose onerous, costly and time-consuming requirements such as additional clinical or animal testing. Regulatory authorities may require that our partners’ change our studies or conduct additional studies, which may significantly delay or make continued pursuit of approval commercially unattractive to our partners. For example, the approval of Baxalta’s HYQVIA BLA was delayed by the FDA until we and Baxalta provided additional preclinical data sufficient to address concerns regarding non-neutralizing antibodies to rHuPH20 that were detected in the registration trial. Although these antibodies have not been associated with any known adverse clinical effects, and the HYQVIA BLA was ultimately approved by the FDA, the FDA or other foreign regulatory agency may, at any time, halt our and our collaborators’ development and commercialization activities due to safety concerns. In addition, even if our product or partners’ products are approved, regulatory agencies may also take post-approval action limiting or revoking our or our partners’ ability to sell these products. Any of these regulatory actions may adversely affect the economic benefit we may derive from our product or our partners’ products and therefore harm our financial condition.
23
Under certain of these regulations, in addition to our partners, we and our contract suppliers and manufacturers are subject to periodic inspection of our or their respective facilities, procedures and operations and/or the testing of products by the FDA, the DEA and other authorities, which conduct periodic inspections to confirm that we and our contract suppliers and manufacturers are in compliance with all applicable regulations. The FDA also conducts pre-approval and post-approval reviews and plant inspections to determine whether our systems, or our contract suppliers’ and manufacturers’ processes, are in compliance with cGMP and other FDA regulations. If our partners, we, or our contract suppliers, fail these inspections, our partners may not be able to commercialize their products in a timely manner without incurring significant additional costs, or at all.
In addition, the FDA imposes a number of complex regulatory requirements on entities that advertise and promote pharmaceuticals including, but not limited to, standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities, and promotional activities involving the Internet.
We may be subject, directly or indirectly, to various broad federal and state healthcare laws. If we are unable to comply, or have not fully complied, with such laws, we could face civil, criminal and administrative penalties, damages, monetary fines, disgorgement, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings and curtailment or restructuring of our operations, any of which could adversely affect our ability to operate.
Our business operations and activities may be directly, or indirectly, subject to various broad federal and state healthcare laws, including without limitation, anti-kickback laws, the Foreign Corrupt Practices Act (FCPA), false claims laws, civil monetary penalty laws, data privacy and security laws, tracing and tracking laws, as well as transparency laws regarding payments or other items of value provided to healthcare providers. These laws may restrict or prohibit a wide range of business activities, including, but not limited to, research, manufacturing, distribution, pricing, discounting, marketing and promotion and other business arrangements. These laws may impact, among other things, our current activities with principal investigators and research subjects, as well as sales, marketing and education programs. Many states have similar healthcare fraud and abuse laws, some of which may be broader in scope and may not be limited to items or services for which payment is made by a government health care program.
Efforts to ensure that our business arrangements will comply with applicable healthcare laws may involve substantial costs. While we have adopted a healthcare corporate compliance program, it is possible that governmental and enforcement authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law interpreting applicable fraud and abuse or other healthcare laws. If our operations or activities are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to, without limitation, civil, criminal and administrative penalties, damages, monetary fines, disgorgement, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings and curtailment or restructuring of our operations, any of which could adversely affect our ability to operate.
In addition, any sales of products outside the U.S. will also likely subject us to the FCPA and foreign equivalents of the healthcare laws mentioned above, among other foreign laws.
We may be required to initiate or defend against legal proceedings related to intellectual property rights, which may result in substantial expense, delay and/or cessation of the development and commercialization of our products.
We primarily rely on patents to protect our intellectual property rights. The strength of this protection, however, is uncertain. For example, it is not certain that:
•we will be able to obtain patent protection for our products and technologies;
•the scope of any of our issued patents will be sufficient to provide commercially significant exclusivity for our products and technologies;
•others will not independently develop similar or alternative technologies or duplicate our technologies and obtain patent protection before we do; and
•any of our issued patents, or patent pending applications that result in issued patents, will be held valid, enforceable and infringed in the event the patents are asserted against others.
24
We currently own or license several patents and also have pending patent applications applicable to rHuPH20 and other proprietary materials. There can be no assurance that our existing patents, or any patents issued to us as a result of our pending patent applications, will provide a basis for commercially viable products, will provide us with any competitive advantages, or will not face third party challenges or be the subject of further proceedings limiting their scope or enforceability. Any weaknesses or limitations in our patent portfolio could have a material adverse effect on our business and financial condition. In addition, if any of our pending patent applications do not result in issued patents, or result in issued patents with narrow or limited claims, this could result in us having no or limited protection against generic or biosimilar competition against our product candidates which would have a material adverse effect on our business and financial condition.
We may become involved in interference proceedings in the U.S. Patent and Trademark Office, or other proceedings in other jurisdictions, to determine the priority, validity or enforceability of our patents. In addition, costly litigation could be necessary to protect our patent position.
We also rely on trademarks to protect the names of our products (e.g. Hylenex recombinant). We may not be able to obtain trademark protection for any proposed product names we select. In addition, product names for pharmaceutical products must be approved by health regulatory authorities such as the FDA in addition to meeting the legal standards required for trademark protection and product names we propose may not be timely approved by regulatory agencies which may delay product launch. In addition, our trademarks may be challenged by others. If we enforce our trademarks against third parties, such enforcement proceedings may be expensive.
We also rely on trade secrets, unpatented proprietary know-how and continuing technological innovation that we seek to protect with confidentiality agreements with employees, consultants and others with whom we discuss our business. Disputes may arise concerning the ownership of intellectual property or the applicability or enforceability of these agreements, and we might not be able to resolve these disputes in our favor.
In addition to protecting our own intellectual property rights, third parties may assert patent, trademark or copyright infringement or other intellectual property claims against us. If we become involved in any intellectual property litigation, we may be required to pay substantial damages, including but not limited to treble damages, attorneys’ fees and costs, for past infringement if it is ultimately determined that our products infringe a third party’s intellectual property rights. Even if infringement claims against us are without merit, defending a lawsuit takes significant time, may be expensive and may divert management’s attention from other business concerns. Further, we may be stopped from developing, manufacturing or selling our products until we obtain a license from the owner of the relevant technology or other intellectual property rights. If such a license is available at all, it may require us to pay substantial royalties or other fees.
Patent protection for protein-based therapeutic products and other biotechnology inventions is subject to a great deal of uncertainty, and if patent laws or the interpretation of patent laws change, our competitors may be able to develop and commercialize products based on our discoveries.
Patent protection for protein-based therapeutic products is highly uncertain and involves complex legal and factual questions. In recent years, there have been significant changes in patent law, including the legal standards that govern the scope of protein and biotechnology patents. Standards for patentability of full-length and partial genes, and their corresponding proteins, are changing. Recent court decisions have made it more difficult to obtain patents, by making it more difficult to satisfy the patentable subject matter requirement and the requirement of non-obviousness, have decreased the availability of injunctions against infringers, and have increased the likelihood of challenging the validity of a patent through a declaratory judgment action. Taken together, these decisions could make it more difficult and costly for us to obtain, license and enforce our patents. In addition, the Leahy-Smith America Invents Act (HR 1249) was signed into law in September 2011, which among other changes to the U.S. patent laws, changes patent priority from “first to invent” to “first to file,” implements a post-grant opposition system for patents and provides for a prior user defense to infringement. These judicial and legislative changes have introduced significant uncertainty in the patent law landscape and may potentially negatively impact our ability to procure, maintain and enforce patents to provide exclusivity for our products.
There also have been, and continue to be, policy discussions concerning the scope of patent protection awarded to biotechnology inventions. Social and political opposition to biotechnology patents may lead to narrower patent protection within the biotechnology industry. Social and political opposition to patents on genes and proteins and recent court decisions concerning patentability of isolated genes may lead to narrower patent protection, or narrower claim interpretation, for isolated genes, their corresponding proteins and inventions related to their use, formulation and manufacture. Patent protection relating to biotechnology products is also subject to a great deal of uncertainty outside the U.S., and patent laws are evolving and undergoing revision in many countries. Changes in, or different interpretations of, patent laws worldwide may result in our inability to obtain or enforce patents, and may allow others to use our discoveries to develop and commercialize competitive products, which would impair our business.
25
If third party reimbursement and customer contracts are not available, Hylenex and our partners’ products may not be accepted in the market resulting in commercial performance below that which was expected or projected.
Our ability to earn sufficient returns on Hylenex and our partners’ ability to earn sufficient returns on their products will depend in part on the extent to which reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers, managed care organizations and other healthcare providers.
Third-party payors are increasingly attempting to limit both the coverage and the level of reimbursement of new drug products to contain costs. Consequently, significant uncertainty exists as to the reimbursement status of newly approved healthcare products. Third party payors may not establish adequate levels of reimbursement for the products that we and our partners commercialize, which could limit their market acceptance and result in a material adverse effect on our revenues and financial condition.
Customer contracts, such as with group purchasing organizations and hospital formularies, will often not offer contract or formulary status without either the lowest price or substantial proven clinical differentiation. If, for example, Hylenex is compared to animal-derived hyaluronidases by these entities, it is possible that neither of these conditions will be met, which could limit market acceptance and result in a material adverse effect on our revenues and financial condition.
The rising cost of healthcare and related pharmaceutical product pricing has led to cost containment pressures from third-party payers as well as changes in federal coverage and reimbursement policies and practices that could cause us and our partners to sell our products at lower prices, and impact access to our and our partners’ products, resulting in less revenue to us.
Any of our proprietary or collaboration products that have been, or in the future are, approved by the FDA may be purchased or reimbursed by state and federal government authorities, private health insurers and other organizations, such as health maintenance organizations and managed care organizations. Such third party payors increasingly challenge pharmaceutical product pricing. The trend toward managed healthcare in the U.S., the growth of such organizations, and various legislative proposals and enactments to reform healthcare and government insurance programs, including the Medicare Prescription Drug Modernization Act of 2003 and the Affordable Care Act of 2010 (ACA), could significantly influence the manner in which pharmaceutical products are prescribed and purchased, resulting in lower prices and/or a reduction in demand. Such cost containment measures and healthcare reforms could adversely affect our ability to sell our product and our partners’ ability to sell their products.
In the U.S., our business may be impacted by changes in federal reimbursement policy resulting from executive actions, federal regulations, or federal demonstration projects.
The federal administration and/or agencies, such as CMS, have announced a number of demonstration projects, recommendations and proposals to implement various elements described in the drug pricing blueprint. CMS, the federal agency responsible for administering Medicare and overseeing state Medicaid programs and Health Insurance Marketplaces, has substantial power to implement policy changes or demonstration projects that can quickly and significantly affect how drugs, including our products, are covered and reimbursed. For example, in November 2020, former President Trump announced the interim final rule to implement the Most Favored Nations drug pricing model seeking to tie Medicare payment rates to an international index price. This final rule is currently subject to a preliminary injunction. Additionally, a number of Congressional committees have also held hearings and evaluated proposed legislation on drug pricing and payment policy which may affect our business. For example, in July 2019, the Senate Finance Committee advanced a bill that in part would penalize pharmaceutical manufacturers for increasing drug list prices covered by Medicare Part B and Part D, faster than the rate of inflation, and cap out-of-pocket expenses for Medicare Part D beneficiaries. Several other proposals have been introduced that, if enacted and implemented, could affect access to and sales of our and our partners’ products, allow the federal government to engage in price negotiations on certain drugs, and allow importation of prescription medication from Canada or other countries.
In this dynamic environment, we are unable to predict which or how many federal policy, legislative or regulatory changes may ultimately be enacted. To the extent federal government initiatives decrease or modify the coverage or reimbursement available for our or our partners’ products, limit or impact our decisions regarding the pricing of biopharmaceutical products or otherwise reduce the use of our or our partners’ U.S. products, such actions could have a material adverse effect on our business and results of operations.
26
Furthermore, individual states are considering proposed legislation and have become increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access, importation from other countries and bulk purchasing. Legally mandated price controls on payment amounts by third party payors or other restrictions could negatively and materially impact our revenues and financial condition. We anticipate that we will encounter similar regulatory and legislative issues in most other countries outside the U.S.
In addition, private payers in the US, including insurers, pharmacy benefit managers (PBMs), integrated healthcare delivery systems, and group purchasing organizations, are continuously seeking ways to reduce drug costs. Many payers have developed and continue to develop ways to shift a greater portion of drug costs to patients through, for example, limited benefit plan designs, high deductible plans and higher co-pay or coinsurance obligations. Consolidation in the payer space has also resulted in a few large PBMs and insurers which place greater pressure on pricing and utilization negotiations for our and our partners’ products in the U.S., increasing the need for higher discounts and rebates and limiting patient access and utilization. Ultimately, additional discounts, rebates and other price reductions, fees, coverage and plan changes, or exclusions imposed by these private payers on our and our partners’ products could have an adverse event on product sales, our business and results of operations.
We also face risks relating to the reporting of pricing data that affects the reimbursement of and discounts provided for our products. Government price reporting regulations are complex and may require a manufacturer to update certain previously submitted data. If our submitted pricing data are incorrect, we may become subject to substantial fines and penalties or other government enforcement actions, which could have a material adverse effect on our business and results of operations. In addition, as a result of restating previously reported price data, we also may be required to pay additional rebates and provide additional discounts.
We face intense competition and rapid technological change that could result in the development of products by others that are competitive with or superior to our proprietary and collaboration products, including those under development.
Our proprietary and collaboration products have numerous competitors in the U.S. and abroad including, among others, major pharmaceutical and specialized biotechnology firms, universities and other research institutions that have developed competing products. Many of these competitors have substantially more resources and product development, manufacturing and marketing experience and capabilities than we do. The competitors for Hylenex recombinant include, but are not limited to, Valeant Pharmaceuticals International, Inc.’s FDA-approved product, Vitrase®, an ovine (ram) hyaluronidase, and Amphastar Pharmaceuticals, Inc.’s product, Amphadase®, a bovine (bull) hyaluronidase. For our ENHANZE technology, such competitors may include major pharmaceutical and specialized biotechnology firms. These competitors may develop technologies and products that are more effective, safer, or less costly than our current or future proprietary and collaboration products and product candidates or that could render our and our partners’ products, technologies and product candidates obsolete or noncompetitive.
General Risks
Our inability to attract, hire and retain key management and scientific personnel could negatively affect our business.
Our success depends on the performance of key management and scientific employees with relevant experience. We depend substantially on our ability to hire, train, motivate and retain high quality personnel, especially our scientists and management team. If we are unable to identify, hire and retain qualified personnel, our ability to support current and future alliances with strategic collaborators could be adversely impacted. Our use of domestic and international third-party contractors, consultants and staffing agencies also subjects us to potential co-employment liability claims.
27
Furthermore, if we were to lose key management personnel, we may lose some portion of our institutional knowledge and technical know-how, potentially causing a disruption or delay in one or more of our partnered development programs until adequate replacement personnel could be hired and trained. In addition, we do not have key person life insurance policies on the lives of any of our employees which would help cover the cost of associated with the loss of key employees.
Our operations might be interrupted by the occurrence of a natural disaster or other catastrophic event.
Our operations, including laboratories, offices and other research facilities, are located in multiple buildings in San Diego, California. We depend on our facilities and on our collaborators, contractors and vendors for the continued operation of our business. Natural disasters or other catastrophic events, pandemics, interruptions in the supply of natural resources, political and governmental changes, wildfires and other fires, floods, explosions, actions of animal rights activists, earthquakes and civil unrest could disrupt our operations or those of our collaborators, contractors and vendors. Even though we believe we carry commercially reasonable business interruption and liability insurance, and our contractors may carry liability insurance that protect us in certain events, we may suffer losses as a result of business interruptions that exceed the coverage available under our and our contractors’ insurance policies or for which we or our contractors do not have coverage. Any natural disaster or catastrophic event could have a significant negative impact on our operations and financial results. Moreover, any such event could delay our partners’ research and development programs.
Cyberattacks, security breaches or system breakdowns may disrupt our operations and harm our operating results and reputation.
We and our partners are subject to increasingly sophisticated attempts to gain unauthorized access to our information technology storage and access systems and are devoting resources to protect against such intrusion. Cyberattacks could render us or our partners unable to utilize key systems or access important data needed to operate our business. The wrongful use, theft, deliberate sabotage or any other type of security breach with respect to any of our or any of our vendors and partners’ information technology storage and access systems could result in the breakdown or other service interruption, or the disruption of our ability to use such systems or disclosure or dissemination of proprietary and confidential information that is electronically stored, including intellectual property, trade secrets, financial information, regulatory information, strategic plans, sales trends and forecasts, litigation materials or personal information belonging to us, our staff, our patients, customers and/or other business partners which could result in a material adverse impact on our business, operating results and financial condition. We continue to invest in monitoring, and other security and data recovery measures to protect our critical and sensitive data and systems. However, these may not be adequate to prevent or fully recover systems or data from all breakdowns, service interruptions, attacks or breaches of our systems. Furthermore, any physical break-in or trespass of our facilities could result in the misappropriation, theft, sabotage or any other type of security breach with respect to our proprietary and confidential information, including research or clinical data or damage to our research and development equipment and assets. Such adverse effects could be material and irrevocable to our business, operating results, financial condition and reputation.
Item 1B.Unresolved Staff Comments
None.
Item 2.Properties
Our administrative offices and research facilities are currently located in San Diego, California. During 2020 we leased an aggregate of approximately 50,000 square feet of office and research space. We believe our facilities are adequate for our current and near-term needs.
Item 3.Legal Proceedings
From time to time, we may be involved in disputes, including litigation, relating to claims arising out of operations in the normal course of our business. Any of these claims could subject us to costly legal expenses and, while we generally believe that we have adequate insurance to cover many different types of liabilities, our insurance carriers may deny coverage or our policy limits may be inadequate to fully satisfy any damage awards or settlements. If this were to happen, the payment of any such awards could have a material adverse effect on our consolidated results of operations and financial position. Additionally, any such claims, whether or not successful, could damage our reputation and business. We currently are not a party to any legal proceedings, the adverse outcome of which, in management’s opinion, individually or in the aggregate, would have a material adverse effect on our consolidated results of operations or financial position.
Item 4.Mine Safety Disclosures
Not applicable.
28
PART II
Item 5.Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information
Our common stock is listed on the NASDAQ Global Select Market under the symbol “HALO.” As of February 17, 2021, we had approximately 52,081 stockholders of record and beneficial owners of our common stock.
Dividends
We have never declared or paid any dividends on our common stock. We currently intend to retain available cash for funding operations and stock repurchases; therefore, we do not expect to pay any dividends on our common stock in the foreseeable future. In addition, the provisions of our borrowing arrangements limit, among other things, our ability to pay dividends and make certain other payments. Any future determination to pay dividends on our common stock will be at the discretion of our board of directors and will depend upon, among other factors, our results of operations, financial condition, capital requirements, contract restrictions, business prospects and other factors our board of directors may deem relevant.
Purchase of Equity Securities by the Issuer
In November, 2019, we announced that the Board of Directors authorized the initiation of a capital return program to repurchase up to $550.0 million of outstanding common stock over a three-year period. During 2019, we repurchased approximately 11.1 million shares of common stock for $200.0 million at an average price of $18.03.
During 2020, we repurchased 6.5 million shares of common stock for $150.0 million at an average price of $23.05. The shares were purchased through open market transactions and through an Accelerated Share Repurchase (ASR) agreement with Bank of America in December 2020, for which we repurchased $21.7 million of common stock and received 0.5 million shares. We retired the repurchased shares and they resumed the status of authorized and unissued shares.
The table below sets forth information regarding repurchases during the three months ended December 31, 2020:
| Period | Total Number of Shares Purchased | Weighted-Average Price paid per share | Total Number of Shares Purchased as Part of Publicly Announced Programs | Approximate Dollar Value of Shares That May Yet Be purchased under the Programs | ||||||||||||||||||||||
| October 1, 2020 through October 31, 2020 | 531,674 | $ | 27.69 | 531.674 | $ | 222,798 | ||||||||||||||||||||
| November 1, 2020 through November 30, 2020 | 43,247 | $ | 27.79 | 43.247 | $ | 221,595 | ||||||||||||||||||||
December 1, 2020 through December 31, 2020 (1) | 520,445 | $ | 41.71 | 520.445 | $ | 199,885 | ||||||||||||||||||||
| Total | 1,095,366 | 1,095.366 | ||||||||||||||||||||||||
(1) These shares were repurchased through the December 2020 ASR.
29
Stock Performance Graph and Cumulative Total Return
Notwithstanding any statement to the contrary in any of our previous or future filings with the SEC, the following information relating to the price performance of our common stock shall not be deemed to be “filed” with the SEC or to be “soliciting material” under the Securities Exchange Act of 1934, as amended, or the Exchange Act, and it shall not be deemed to be incorporated by reference into any of our filings under the Securities Act of 1933, as amended, or the Exchange Act, except to the extent we specifically incorporate it by reference into such filing.
The graph below compares Halozyme Therapeutics, Inc.’s cumulative five-year total shareholder return on common stock with the cumulative total returns of the NASDAQ Composite Index and the NASDAQ Biotechnology Index. The graph tracks the performance of a $100 investment in our common stock and in each of the indexes (with the reinvestment of all dividends) from December 31, 2015 to December 31, 2020. The historical stock price performance included in this graph is not necessarily indicative of future stock price performance.
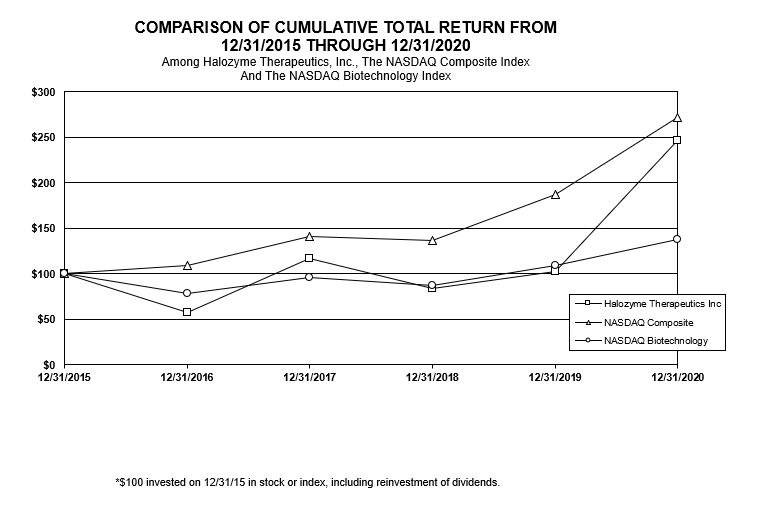
| 12/31/2015 | 12/31/2016 | 12/31/2017 | 12/31/2018 | 12/31/2019 | 12/31/2020 | |||||||||||||||
| Halozyme Therapeutics, Inc. | $100 | $57 | $117 | $84 | $102 | $246 | ||||||||||||||
| NASDAQ Composite | $100 | $109 | $141 | $137 | $187 | $272 | ||||||||||||||
| NASDAQ Biotechnology | $100 | $79 | $96 | $87 | $109 | $138 | ||||||||||||||
30
Item 6.Selected Financial Data
The selected consolidated financial data set forth below as of December 31, 2020 and 2019, and for the years ended December 31, 2020, 2019 and 2018, are derived from our audited consolidated financial statements included elsewhere in this report. This information should be read in conjunction with those consolidated financial statements, the notes thereto, and with “Management’s Discussion and Analysis of Financial Condition and Results of Operations.” The selected consolidated financial data set forth below as of December 31, 2018, 2017 and 2016, and for the years ended December 31, 2017 and 2016, are derived from our audited consolidated financial statements that are contained in reports previously filed with the SEC, not included herein.
Summary Financial Information
| Year Ended December 31, | ||||||||||||||||||||||||||||||||
| Statement of Operations Data: | 2020 | 2019 | 2018 | 2017 | 2016 | |||||||||||||||||||||||||||
| (in thousands, except for per share amounts) | ||||||||||||||||||||||||||||||||
| Total revenues | $ | 267,594 | $ | 195,992 | $ | 151,862 | $ | 316,613 | $ | 146,691 | ||||||||||||||||||||||
| Net income (loss) | $ | 129,085 | $ | (72,240) | $ | (80,330) | $ | 62,971 | $ | (103,023) | ||||||||||||||||||||||
| Net income (loss) per share, basic | $ | 0.95 | $ | (0.50) | $ | (0.56) | $ | 0.46 | $ | (0.81) | ||||||||||||||||||||||
| Net income (loss) per share, diluted | $ | 0.91 | $ | (0.50) | $ | (0.56) | $ | 0.45 | $ | (0.81) | ||||||||||||||||||||||
| Shares used in computing net income (loss) per share, basic | 136,206 | 144,329 | 143,599 | 136,419 | 127,964 | |||||||||||||||||||||||||||
| Shares used in computing net income (loss) per share, diluted | 141,463 | 144,329 | 143,599 | 139,068 | 127,964 | |||||||||||||||||||||||||||
| As of December 31, | ||||||||||||||||||||||||||||||||
| Balance Sheet Data: | 2020 | 2019 | 2018 | 2017 | 2016 | |||||||||||||||||||||||||||
| (in thousands) | ||||||||||||||||||||||||||||||||
| Cash and cash equivalents and available-for-sale marketable securities | $ | 368,013 | $ | 421,262 | $ | 354,526 | $ | 469,214 | $ | 204,981 | ||||||||||||||||||||||
| Working capital | $ | 133,379 | $ | 457,799 | $ | 278,488 | $ | 379,044 | $ | 201,947 | ||||||||||||||||||||||
| Total assets | $ | 579,924 | $ | 565,874 | $ | 440,248 | $ | 519,945 | $ | 261,515 | ||||||||||||||||||||||
| Deferred revenue | $ | 5,772 | $ | 5,259 | $ | 9,255 | $ | 60,865 | $ | 44,618 | ||||||||||||||||||||||
| Long-term debt, net | $ | — | $ | 383,045 | $ | 34,874 | $ | 125,140 | $ | 199,228 | ||||||||||||||||||||||
| Total liabilities | $ | 428,877 | $ | 474,109 | $ | 191,361 | $ | 311,579 | $ | 293,996 | ||||||||||||||||||||||
| Stockholders’ equity (deficit) | $ | 151,047 | $ | 91,765 | $ | 248,887 | $ | 208,366 | $ | (32,481) | ||||||||||||||||||||||
31
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
In addition to historical information, the following discussion contains forward-looking statements that are subject to risks and uncertainties. Actual results may differ substantially from those referred to herein due to a number of factors, including but not limited to risks described in the Part I, Item 1A, Risks Factors, and elsewhere in this Annual Report. References to “Notes” are Notes included in our Notes to Consolidated Financial Statements.
Overview
Halozyme Therapeutics, Inc. is a biopharma technology platform company that provides innovative and disruptive solutions with the goal of improving patient experience and outcomes. Our proprietary enzyme, rHuPH20, is used to facilitate the delivery of injected drugs and fluids. We license our technology to biopharmaceutical companies to collaboratively develop products that combine our ENHANZE® drug delivery technology with the collaborators’ proprietary compounds.
Our approved product and our collaborators’ approved products and product candidates are based on rHuPH20, our patented recombinant human hyaluronidase enzyme. rHuPH20 is the active ingredient in our first commercially approved product, Hylenex® recombinant, and it works by breaking down hyaluronan (or HA), a naturally occurring carbohydrate that is a major component of the extracellular matrix in tissues throughout the body such as skin and cartilage. This temporarily increases dispersion and absorption allowing for improved subcutaneous delivery of injectable biologics, such as monoclonal antibodies and other large therapeutic molecules, as well as small molecules and fluids. We refer to the application of rHuPH20 to facilitate the delivery of other drugs or fluids as our ENHANZE® drug delivery technology (“ENHANZE”). We license the ENHANZE technology to form collaborations with biopharmaceutical companies that develop or market drugs requiring or benefiting from injection via the subcutaneous route of administration. In the development of proprietary intravenous (IV) drugs combined with our ENHANZE technology, data have been generated supporting the potential for ENHANZE to reduce treatment burden, as a result of shorter duration of subcutaneous (SC) administration. ENHANZE may enable fixed-dose SC dosing compared to weight-based dosing required for IV administration, and potentially allow for lower rates of infusion related reactions. ENHANZE may enable more flexible treatment options such as home administration by a healthcare professional or potentially the patient. Lastly, certain proprietary drugs co-formulated with ENHANZE have been granted additional exclusivity, extending the patent life of the product beyond the one of the proprietary IV drug.
We currently have ENHANZE collaborations with F. Hoffmann-La Roche, Ltd. and Hoffmann-La Roche, Inc. (Roche), Baxalta US Inc. and Baxalta GmbH (now members of the Takeda group of companies, following the acquisition of Shire plc by Takeda Pharmaceutical Company Limited in January 2019) (Baxalta), Pfizer Inc. (Pfizer), Janssen Biotech, Inc. (Janssen), AbbVie, Inc. (AbbVie), Eli Lilly and Company (Lilly), Bristol-Myers Squibb Company (BMS), Alexion Pharma Holding (Alexion), ARGENX BVBA (argenx) and Horizon Therapeutics plc. (Horizon). We receive royalties from three of these collaborations, including royalties from sales of one product from the Baxalta collaboration, three products from the Roche collaboration and one product from Janssen collaboration. Future potential revenues from royalties and fees from ENHANZE collaborations and the sales and/or royalties of our approved products will depend on the ability of Halozyme and our collaborators to develop, manufacture, secure and maintain regulatory approvals for approved products and product candidates and commercialize product candidates.
32
Our 2020 and recent key events are as follows:
Janssen
•In January 2021, we announced that Janssen received accelerated approval from the FDA for DARZALEX FASPRO in combination with bortezomib, cyclophosphamide and dexamethasone (D-VCd) for the treatment of adult patients with newly diagnosed light chain (AL) amyloidosis. AL amyloidosis is a rare and potentially fatal disease that develops when plasma cells in the bone marrow generate abnormal light chains, which form amyloid deposits in vital organs and lead to organ deterioration. There were no approved therapies for these patients prior to Darzalex FASPRO.
•In December 2020, Janssen achieved the first sales milestone for Darzalex FASPRO/Darxalex SC, triggering a payment of $15.0 million.
•In November 2020, Janssen initiated a Phase 1 study of amivantamab using ENHANZE technology.
•In November 2020, we announced that Janssen submitted regulatory applications to the FDA and EMA seeking approval of the DARZALEX FASPRO in the U.S. and as DARZALEX SC in the European Union (EU) in combination with pomalidomide and dexamethasone (D-Pd) for the treatment of patients with relapsed or refractory multiple myeloma who have received at least one prior line of therapy.
•In November 2020, Janssen announced it submitted a Type II variation application to the EMA seeking European approval for the DARZALEX SC to be used in the treatment of patients with AL amyloidosis.
•In August 2020, Janssen announced that Health Canada approved DARZALEX SC in four regimens across five indications in patients with multiple myeloma, most notably newly diagnosed, transplant-ineligible patients as well as relapsed or refractory patients.
•In June 2020, we announced that Janssen received European marketing authorization and launched the commercial sale of DARZALEX SC in the European Union, triggering a $10.0 million milestone payment. The approval applies to all current daratumumab indications in frontline and relapsed/refractory settings.
•In May 2020, we announced that Janssen received US FDA approval of DARZALEX FASPRO in four of six regimens approved for the intravenous form in multiple myeloma patients, including newly diagnosed, transplant-ineligible patients as well as relapsed or refractory patients and launched the commercial sale, triggering a $15 million milestone payment.
•In April 2020, we announced the submission of a New Drug Application (NDA) to Japan's Ministry of Health, Labour and Welfare (MHLW) by Janssen seeking approval of a new SC formulation of daratumumab, an intravenous (IV) treatment approved for patients with multiple myeloma.
Argenx
•In January 2021, argenx initiated a Phase 3 study of ARGX-113 utilizing ENHANZE in pemphigus vulgaris (PV), a rare autoimmune disease that causes painful blisters on the skin and mucous membranes.
•In December 2020, argenx initiated a Phase 3 study of ARGX-113 using ENHANZE technology for patients with immune thrombocytopenia (ITP), an immune disorder in which the blood does not clot normally, triggering a $15.0 million payment.
•In October 2020, we announced that argenx expanded the existing global collaboration and license agreement that was signed in February 2019. Under the newly announced expansion, argenx gained the ability to exclusively access our ENHANZE technology for three additional targets upon nomination, for a total of up to six targets. To date, argenx has nominated two targets including the human neonatal Fc receptor FcRn and complement component C2.
Roche
•In December 2020, Roche initiated a Phase 3 study in patients with non-small cell lung cancer for Tecentriq using ENHANZE technology, triggering a $17.0 million payment.
33
•In December 2020, we announced that the European Commission approved Roche's Phesgo, a fixed-dose combination of Perjeta (pertuzumab) and Herceptin (trastuzumab) with ENHANZE technology, administered by subcutaneous injection for the treatment of patients with early and metastatic HER2-positive breast cancer. This is the first time the European Commission has approved a product combining two monoclonal antibodies that can be administered by a single subcutaneous injection utilizing ENHANZE technology.
•In June 2020, we announced that Roche received FDA approval of Phesgo utilizing ENHANZE technology for the treatment of patients with HER2-positive breast cancer.
Horizon
•In November 2020, we announced a global collaboration and license agreement that gives Horizon exclusive access to ENHANZE technology for SC formulation of medicines targeting IGF-1R. Horizon intends to use ENHANZE to develop a SC formulation of TEPEZZA (teprotumumab-trbw), indicated for the treatment of thyroid eye disease, a serious, progressive and vision-threatening rare autoimmune disease, potentially shortening drug administration time, reducing healthcare practitioner time and offering additional flexibility and convenience for patients.
Baxalta
•In September 2020, Takeda announced that the EMA approved a label update for HYQVIA broadening its use and making it the first and only facilitated subcutaneous immunoglobulin replacement therapy in adults, adolescents and children with an expanded range of secondary immunodeficiencies (SID).
BMS
•In June 2020, BMS selected 3 targets on an exclusive basis and exercised their option to convert a co-exclusive license to an exclusive license, triggering a $5.0 million payment. BMS has selected eight targets on an exclusive basis to date.
Corporate
•In March 2020, Hylenex, which was approved in an NDA, was deemed to be an approved Biologics License Application (BLA) under section 351(a) of the Public Health Service Act (PHS Act). A portion of our competition comes from compounding pharmacies, which are prohibited from compounding biologic products per the Drug Quality & Security Act (DQSA). As hyaluronidase is now considered a biologic, this change may benefit sales of Hylenex.
•During 2020 we repurchased 6.5 million shares of common stock in open market and accelerated share repurchases for $150.0 million at an average price per share of $23.05. Since the inception of our capital return program in November 2019 to repurchase up to $550.0 million of outstanding common stock over a three-year period, we have repurchased a total of 17.6 million shares for $350.0 million at an average price per share of $19.88 as of December 31, 2020.
34
Results of Operations
Comparison of Years Ended December 31, 2020 and 2019
Royalties – Royalty revenue was $88.6 million in 2020 compared to $69.9 million in 2019. The increase was mainly driven by sales of DARZALEX FASPRO in the U.S and EU by Janssen due to the product launch in the second quarter of 2020, partially offset by slightly lower sales of Herceptin SC and MabThera SC by Roche. In general, we expect royalty revenue to grow as a result of our recent ENHANZE partner product launches, offsetting the ongoing impact from biosimilars in Europe related to our mature ENHANZE partner products.
Product Sales, Net – Product sales, net were as follows (in thousands):
| Year Ended December 31, | |||||||||||||||||||||||
| 2020 | 2019 | Dollar Change | Percentage Change | ||||||||||||||||||||
| Sales of bulk rHuPH20 | $ | 38,237 | $ | 48,285 | $ | (10,048) | (21) | % | |||||||||||||||
| Sales of ENHANZE drug product | 719 | 768 | (49) | (6) | % | ||||||||||||||||||
| Sales of Hylenex | 17,031 | 16,995 | 36 | — | % | ||||||||||||||||||
| Total product sales, net | $ | 55,987 | $ | 66,048 | $ | (10,061) | (15) | % | |||||||||||||||
Product sales, net was lower by $10.1 million in 2020 compared to 2019, due to lower sales of rHuPH20 as a result of a one-off large partner order in 2019. We expect that product sales of bulk rHuPH20 and ENHANZE drug product will fluctuate in future periods based on the needs of our collaborators. In March 2020, the Surgeon General advised hospitals to cancel elective surgeries due to COVID-19. Hylenex is used in cataract surgery and other ophthalmologic surgeries, and therefore the advisory resulted in a decrease in sales of Hylenex in the second quarter of 2020. Most states resumed elective surgeries in April and May of 2020 which supported an increase in sales in the second half of 2020.
Revenues Under Collaborative Agreements – Revenues under collaborative agreements were as follows (in thousands):
| Year Ended December 31, | |||||||||||||||||||||||
| 2020 | 2019 | Dollar Change | Percentage Change | ||||||||||||||||||||
| Upfront license fees, license fees for the election of additional targets, event-based payments, license maintenance fees and amortization of deferred upfront and other license fees: | |||||||||||||||||||||||
| Janssen | $ | 42,000 | $ | — | $ | 42,000 | 100 | % | |||||||||||||||
| Horizon | 30,000 | — | 30,000 | 100 | % | ||||||||||||||||||
| Roche | 17,000 | 10,000 | 7,000 | 70 | % | ||||||||||||||||||
| argenx | 20,000 | 45,000 | (25,000) | (56) | % | ||||||||||||||||||
| BMS | 12,264 | — | 12,264 | 100 | % | ||||||||||||||||||
| Other | 500 | 3,500 | (3,000) | (86) | % | ||||||||||||||||||
| Subtotal | 121,764 | 58,500 | 63,264 | 108 | % | ||||||||||||||||||
| Reimbursements for research and development services: | 1,247 | 1,545 | (298) | (19) | % | ||||||||||||||||||
| Total revenues under collaborative agreements | $ | 123,011 | $ | 60,045 | $ | 62,966 | 105 | % | |||||||||||||||
Revenue from license fees increased $63.3 million in 2020, compared to 2019 mainly due to the recognition of $121.3 million related to the Janssen, Horizon, Roche, argenx and BMS collaborations in the current year, as compared to $55.0 million recognized in connection with argenx and Roche collaboration in 2019. Revenue from upfront licenses fees, license fees for the election of additional targets, license maintenance fees and other license fees and event-based payments vary from period to period based on our ENHANZE collaboration activity. We expect these revenues to continue to fluctuate in future periods based on our collaborators’ ability to meet various clinical and regulatory milestones set forth in such agreements and
35
our ability to obtain new collaborative agreements. To date, based on current partner communications, we have not identified any COVID-19 related delays that will materially impact our ENHANZE milestone revenue. We will continue to monitor the ENHANZE development activities of our partners and the impact of their plans on our milestone revenue, as there may be potential for COVID-19 related delays.
Cost of Product Sales –Cost of product sales were $43.4 million in 2020 compared to $45.5 million in 2019. The decrease of $2.1 million in cost of product sales was mainly due to a decrease in sales of bulk rHuPH20 to Janssen, partially offset by an increase in sales of bulk rHuPH20 to Baxalta and Roche.
Research and Development – Research and development expenses consist of external costs, salaries and benefits and allocation of facilities and other overhead expenses related to research manufacturing, clinical trials, preclinical and regulatory activities. Research and development expenses incurred were as follows (in thousands):
| Year Ended December 31, | |||||||||||||||||||||||
| 2020 | 2019 | Dollar Change | Percentage Change | ||||||||||||||||||||
| Programs | |||||||||||||||||||||||
| PEGPH20 | $ | 7,201 | $ | 103,150 | $ | (95,949) | (93) | % | |||||||||||||||
| Restructuring | — | 17,201 | (17,201) | (100) | % | ||||||||||||||||||
| ENHANZE collaborations and rHuPH20 platform | 26,002 | 18,896 | 7,106 | 38 | % | ||||||||||||||||||
| Other | 1,033 | 1,584 | (551) | (35) | % | ||||||||||||||||||
| Total research and development expenses | $ | 34,236 | $ | 140,831 | $ | (106,595) | (76) | % | |||||||||||||||
Research and development expenses relating to our PEGPH20 programs for 2020 decreased by 93%, compared to the same period in 2019, primarily due to decreased clinical trial activities. On November 4, 2019, we announced that the HALO-301 clinical study failed to reach the primary endpoint of overall survival. As a result, we halted development activities for PEGPH20, closed our oncology operations and began the close out process for all our clinical trials. We implemented an organizational restructuring to focus our operations solely on ENHANZE, which was completed in the second quarter of 2020 and resulted in a reduction in research and development expenses.
Research and development expenses relating to our ENHANZE collaborations and our rHuPH20 platform for 2020 increased by 38%, compared to 2019, primarily due to increased costs to support additional ENHANZE targets and a higher allocation of overhead costs. The rHuPH20 platform includes research and development expenses related to our proprietary rHuPH20 enzyme that were not designated to a specific program at the time the expenses were incurred.
Selling, General and Administrative – Selling, general and administrative (SG&A) expenses were $45.7 million in 2020 compared to $77.3 million in 2019. The decrease of $31.6 million, or 41%, was primarily due to a decrease in compensation expense, one time restructuring cost in the prior year and discontinuation of commercial expenses related to market research and educational activities as we prepared for a potential commercial launch of PEGPH20. The discontinuation of our development activities for PEGPH20 and closure of our oncology operations resulted in a reduction in commercialization activities and compensation expense.
Interest Expense – Interest expense was $20.4 million in 2020 compared to $11.6 million in 2019. The increase of $8.8 million was primarily due to interest expense related to the Convertible Notes, offset by a discontinuation in interest expense for the Royalty-backed Loan and the Oxford and SVB Loan following the repayment of those obligations. We expect interest expense related to the Convertible Notes to decrease by approximately $12.0 million in 2021 as the non-cash interest expense associated with the amortization of the equity component of Convertible Notes will be eliminated upon the adoption of ASU 2020-06.
36
Comparison of Years Ended December 31, 2019 and 2018
For discussion related to changes in financial condition and the results of operations for fiscal year 2019 compared to fiscal year 2018, refer to Part II - Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations included in the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2019, which was filed with the SEC on February 24, 2020.
Liquidity and Capital Resources
Our principal sources of liquidity are our existing cash, cash equivalents and available-for-sale marketable securities. As of December 31, 2020, we had cash, cash equivalents and marketable securities of $368.0 million. We believe that our current cash, cash equivalents and marketable securities will be sufficient to fund our operations for at least the next twelve months. We expect to fund our operations going forward with existing cash resources, anticipated revenues from our existing collaborations and cash that we may raise through future transactions. We may raise cash through any one of the following financing vehicles: (i) new collaborative agreements; (ii) expansions or revisions to existing collaborative relationships; (iii) private financings; (iv) other equity or debt financings; (v) monetizing assets; and/or (vi) the public offering of securities.
We may, in the future, offer and sell additional equity, debt securities and warrants to purchase any of such securities, either individually or in units to raise capital to raise funds for additional working capital, capital expenditures, share repurchases, acquisitions or for other general corporate purposes.
Cash Flows
Operating Activities
Net cash provided by operations was $55.5 million in 2020 compared to cash used in by operation $85.4 million in 2019. The $140.9 million increase in cash provided by operations was mainly due to an increase in cash received related to collaboration partner license of $119.0 million during the current year, offset by $55.0 million owed from argenx and Roche for license fees in the prior year, and an increase in working capital spend for 2020 compared to the prior year.
Investing Activities
Net cash provided by investing activities was $78.4 million in 2020 compared to net cash used in by investing activities $5.5 million in 2019. The increase in net cash provided by investing activities was primarily due to the increase in proceeds from maturities of marketable securities for 2020.
Financing Activities
Net cash used in financing activities was $106.3 million in 2020, compared to net cash provided by financing activities of $153.2 million in 2019, mainly due to a decrease in cash proceeds from issuance of long-term debt, offset by a decrease of $49.9 million in repurchase of common stock, a $49.2 million increase in net proceeds from the issuance of common stock under equity incentive plans and a decrease in the amount of repayments of long-term debt of $88.5 million in 2020.
Share Repurchases
The Board of Directors approved a share repurchase program, pursuant to which we may repurchase our issued and outstanding shares of common stock from time to time. The Company retired the repurchased shares. See Note 8. Stockholders’ Equity, within the notes to the consolidated financial statements for additional information regarding our share repurchases.
37
Debt
Convertible Notes
In November 2019, we completed the sale of $460.0 million in aggregate principal amount of 1.25% Convertible Senior Notes due in 2024 (Convertible Notes) in a private placement to qualified institutional buyers. We received net proceeds from the offering of approximately $447.4 million. We used $200.0 million of the net proceeds from the offering to repurchase shares of our common stock, including approximately $143.1 million to repurchase approximately 8.1 million shares of common stock concurrently with the offering in privately negotiated transactions, $6.9 million in open market purchases and $50.0 million to repurchase approximately 2.6 million shares of common stock through an accelerated share repurchase agreement.
We used approximately $26.1 million of the net proceeds from the offering to repay all outstanding amounts under our loan agreement with Oxford Finance and Silicon Valley Bank and intend to use the remainder of the net proceeds for general corporate purposes, including additional share repurchases subsequent to the offering, and working capital.
The Convertible Notes will pay interest semi-annually in arrears on June 1st and December 1st of each year, beginning on June 1, 2020, at an annual rate of 1.25% and will be convertible into cash, shares of common stock or a combination of cash and shares of common stock, at our election, based on the applicable conversion rate at such time. The Convertible Notes are general unsecured obligations and will rank senior in right of payment to all indebtedness that is expressly subordinated in right of payment to the Convertible Notes, will rank equally in right of payment with all existing and future liabilities that are not so subordinated, will be effectively junior to any secured indebtedness to the extent of the value of the assets securing such indebtedness and will be structurally subordinated to all indebtedness and other liabilities (including trade payables) of our current or future subsidiaries.
Holders may convert their Convertible Notes at their option only in the following circumstances: (1) during any calendar quarter commencing after the calendar quarter ending on March 31, 2020, if the last reported sale price per share of common stock exceeds 130% of the conversion price for each of at least 20 trading days during the 30 consecutive trading days ending on, and including, the last trading day of the immediately preceding calendar quarter; (2) during the five consecutive business days immediately after any five consecutive trading day period (such five consecutive trading day period, the “measurement period”) in which the trading price per $1,000 principal amount of notes for each trading day of the measurement period was less than 98% of the product of the last reported sale price per share of Company’s common stock on such trading day and the conversion rate on such trading day; (3) upon the occurrence of certain corporate events or distributions on Company’s common stock, as described in the offering memorandum; (4) if we call such notes for redemption; and (5) at any time from, and including, June 1, 2024 until the close of business on the scheduled trading day immediately before the maturity date of December 1, 2024. The Convertible Notes will be convertible, regardless of the foregoing circumstances, at any time from, and including, June 1, 2024 until the close of business on the scheduled trading day immediately preceding the maturity date.
In January 2021, we notified the note holders that we will settle the principal of the Convertible Notes in cash. Therefore, upon conversion, the principal value of the Convertible Notes will be paid in cash and depending on our stock price, any additional amount over principal amount will be settled in shares of common stock. The initial conversion rate for the Convertible Notes will be 41.9208 shares of common stock per $1,000 in principal amount of Convertible Notes, equivalent to a conversion price of approximately $23.85 per share of our common stock. The conversion rate is subject to adjustment as described in the Indenture.
Upon the occurrence of certain circumstances, holders of the Convertible Notes may require us to purchase all or portion of their notes for cash, which may require the use of a substantial amount of cash. As of December 31, 2020, the conditional conversion feature was triggered and our notes are classified as a current liability. At December 31, 2020, we had a working capital position of $133.4 million, which includes the Convertible Notes that are currently redeemable as of December 31, 2020 but excludes another $65.6 million that is classified as equity. The debt may change from current to non-current period over period, primarily as a result of changes in our stock price. We believe that it is remote that holders of the notes would choose to convert their notes early because the fair value of the security that a note holder can currently realize in an active market is greater than the conversion value the note holder would realize upon early conversion. In the unlikely event that all the debt was converted, we will have two business days following a forty trading day observation period from the conversion date to pay the
38
principal in cash. For the year ended December 31, 2020, we have positive operating income and positive cash flow from operations and, accordingly, while there can be no assurance, we believe we have the ability to generate sufficient cash flows from operations or to raise additional capital through a variety of financing arrangements to satisfy early conversion of the Convertible Notes.
Royalty-backed Loan
In January 2016, through our wholly-owned subsidiary Halozyme Royalty LLC (Halozyme Royalty), we received a $150 million loan (the Royalty-backed Loan) pursuant to a credit agreement (the Credit Agreement) with BioPharma Credit Investments IV Sub, LP and Athyrium Opportunities II Acquisition LP (the Royalty-backed Lenders). Under the terms of the Credit Agreement, Halozyme Therapeutics, Inc. transferred to Halozyme Royalty the right to receive royalty payments from the commercial sales of ENHANZE products owed under the Roche Collaboration and Baxalta Collaboration (Collaboration Agreements). The royalty payments from the Collaboration Agreements were used to repay the principal and interest on the loan (the Royalty Payments). The Royalty-backed Loan bore interest at a per annum rate of 8.75% plus the three-month LIBOR rate. The three-month LIBOR rate was subject to a floor of 0.7% and a cap of 1.5%. In June 2020, we paid the full remaining balance and final payment of $2.93 million thereby satisfying and discharging all obligations under, and terminating, the Royalty-backed Loan.
Oxford and SVB Loan and Security Agreement
In June 2016, we entered into a Loan and Security Agreement (the Loan Agreement) with Oxford Finance LLC (Oxford) and Silicon Valley Bank (SVB) (collectively, the Lenders), providing a senior secured loan facility of up to an aggregate principal amount of $70.0 million, comprising a $55.0 million draw in June 2016 and an additional $15.0 million tranche, which we had the option to draw during the second quarter of 2017 and did not exercise. The proceeds were partially used to pay the outstanding principal and final payment owed on a previous loan agreement with the Lenders. The remaining proceeds were used for working capital and general business requirements. The Loan Agreement repayment schedule provided for interest only payments for the first 18 months, followed by consecutive equal monthly payments of principal and interest in arrears through the maturity date of January 1, 2021. The Loan Agreement provided for a final payment equal to 5.50% of the initial $55 million principal amount. The final payment was due when the Loan Agreement becomes due or upon the prepayment of the facility. We had the option to prepay the outstanding balance of the Loan Agreement in full and exercised this option in November 2019, at which point we paid the full remaining balance and final payment of $26.1 million, thereby satisfying and discharging all obligations under, and terminating, the Loan Agreement.
Off-Balance Sheet Arrangements
As of December 31, 2020, we did not have any relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities, which would have been established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes. In addition, we did not engage in trading activities involving non-exchange traded contracts. As such, we are not materially exposed to any financing, liquidity, market or credit risk that could arise if we had engaged in such relationships.
39
Contractual Obligations
As of December 31, 2020, future minimum payments due under our contractual obligations are as follows (in thousands):
| Payments Due by Period | ||||||||||||||||||||||||||||||||
| Contractual Obligations | Total | Less than 1 Year | 1-3 Years | 4-5 Years | More than 5 Years | |||||||||||||||||||||||||||
Short-term debt(1) | $ | 460,000 | $ | 460,000 | $ | — | $ | — | $ | — | ||||||||||||||||||||||
Interest on short-term debt(2) | 5,750 | 5,750 | — | — | — | |||||||||||||||||||||||||||
Operating leases(3) | 5,277 | 2,563 | 2,714 | — | — | |||||||||||||||||||||||||||
Third-party manufacturing obligations(4) | 77,530 | 46,884 | 30,646 | — | — | |||||||||||||||||||||||||||
| Purchase obligations | 1,168 | 524 | 644 | — | — | |||||||||||||||||||||||||||
| Total | $ | 549,725 | $ | 515,721 | $ | 34,004 | $ | — | $ | — | ||||||||||||||||||||||
_______________
(1).Short-term debt consists of the Convertible Notes.
(2).Interest on short-term debt includes semi-annual interest payments on the Convertible Note. The Convertible Note bears interest at an annual rate of 1.25%.
(3).Includes minimum lease payments related to leases of our office and research facilities and certain autos under non-cancelable operating leases.
(4).We have contracted with third-party manufacturers for the supply of bulk rHuPH20 and fill/finish of Hylenex recombinant. Under these agreements, we are required to purchase certain quantities each year during the terms of the agreements. The amounts presented represent our estimates of the minimum required payments under these agreements.
Contractual obligations for purchases of goods or services include agreements that are enforceable and legally binding to us and that specify all significant terms, including fixed or minimum quantities to be purchased; fixed, minimum or variable price provisions; and the approximate timing of the transaction. For obligations with cancellation provisions, the amounts included in the preceding table were limited to the non-cancelable portion of the agreement terms or the minimum cancellation fee.
For certain restricted stock units and performance stock units granted, the number of shares issued on the date the restricted stock units vest is net of the minimum statutory withholding requirements that we pay in cash to the appropriate taxing authorities on behalf of our employees. The obligation to pay the relevant taxing authority is not included in the preceding table, as the amount is contingent upon continued employment. In addition, the amount of the obligation is unknown, as it is based in part on the market price of our common stock when the awards vest.
The expected timing of payments of the obligations above is estimated based on current information. Timing of payments and actual amounts paid may be different, depending on the time of receipt of goods or services, or changes to agreed-upon amounts for some obligations.
40
Our future capital uses and requirements depend on numerous forward-looking factors. These factors may include, but are not limited to, the following:
•the rate of progress and cost of research and development activities;
•the number and scope of our research and development activities;
•the costs of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights;
•our ability to establish and maintain product discovery and development collaborations, including scale-up manufacturing costs for our collaborators’ product candidates;
•the amount of royalties and milestones from our collaborators;
•the amount of product sales for Hylenex recombinant;
•the costs of obtaining and validating additional manufacturers of rHuPH20;
•the effect of competing technological and market developments;
•the terms and timing of any collaborative, licensing and other arrangements that we may establish; and
•the extent to which we acquire or in-license new products, technologies or businesses.
41
Critical Accounting Estimates
The discussion and analysis of our financial condition and results of operations are based on our consolidated financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles, or U.S. GAAP. The preparation of our consolidated financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses and related disclosure of contingent assets and liabilities. We review our estimates on an ongoing basis. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities. Actual results may differ from these estimates under different assumptions or conditions. Our significant accounting policies are outlined in Note 2 to the Consolidated Financial Statements included in the Form 10-K. We believe the following accounting policies to be critical to the judgments and estimates used in the preparation of our consolidated financial statements.
| Revenue Recognition | ||||||||||||||
| Methodology | Judgment and Uncertainties | Effect if Actual Results Differ From Assumptions | ||||||||||||
| For collaborative agreements, we are entitled to receive event-based payments subject to the collaboration partner's achievement of specified development and regulatory milestones. We recognize revenue when it is deemed probable that these milestones will be achieved, which could be in a period prior to its actual occurrence. At the end of each reporting period, we re-evaluate the probability of achievement of such milestones, and if necessary, adjust our estimate of the overall transaction price. | Revenue is recognized when we determine it is probable a milestone will be achieved. This assessment is based on our past experience with our collaboration partners, market insight and partner communication. | A revenue reversal will be required in the event it is determined that achievement of a milestone, previously deemed probable, will not occur. This reversal may be material. | ||||||||||||
| For collaborative agreements, royalty revenue is recognized in the period the underlying sales occur, but we do not receive final royalty reports from our collaboration partners until after we complete our financial statements for a prior quarter. Therefore, we recognize revenue based on estimates of the royalty earned, which are based on preliminary reports provided by our collaboration partners. | The amount of royalty revenue recognized for the quarter is estimated using our knowledge of past royalty payments, market insight and an estimate made by our collaboration partners provided in a preliminary report. | A final royalty report and associated royalty payment is received approximately 60 days after quarter-end. If necessary, a true-up is recorded at that time if there is a difference from the initial estimated royalty revenue recorded. To date, the true-up entries have not been material. | ||||||||||||
| For collaborative arrangements, when necessary, we perform an allocation of the upfront amount based on relative stand-alone selling prices (SSP) of licenses for individual targets. We determine license SSP using an income-based valuation approach utilizing risk-adjusted discounted cash flow projections. | The inputs used in the valuation model to determine SSP are based on estimates utilizing market data and information provided by our collaboration partners. | Differences in the allocation of the transaction price between delivered and undelivered performance obligations can impact the timing of revenue recognition but do not change the total revenue recognized under any agreement. | ||||||||||||
42
| Share-Based Payments | ||||||||||||||
| Methodology | Judgment and Uncertainties | Effect if Actual Results Differ From Assumptions | ||||||||||||
| We maintain a Stock Incentive Plan, which provides for share-based payment awards, including stock options, restricted stock and performance awards. We determine the fair value of our stock option awards and performance awards at the date of grant using a Black-Scholes model. We determine the fair value of our restricted stock awards at the date of grant using the closing market value of our common stock on the date of grant. | Option-pricing models and generally accepted valuation techniques require management to make assumptions and to apply judgment to determine the fair value of our awards. These assumptions and judgments include estimating the future volatility of our stock price, expected dividend yield and future employee stock option exercise behaviors. Changes in these assumptions can materially affect the fair value estimate. Our performance awards require management to make assumptions regarding the likelihood of achieving long-term Company goals. | We do not currently believe there is a reasonable likelihood that there will be a material change in estimates or assumptions we use to determine stock-based compensation expense. However, if actual results are not consistent with our estimates or assumptions, we may be exposed to changes in share-based compensation expense that could be material. If actual results are not consistent with the assumptions used, the share-based compensation expense reported in our financial statements may not be representative of the actual economic cost of the share-based compensation. A 10% change in our share-based compensation expense for the year ended December 31, 2020 would have affected pre-tax earnings by approximately $1.7 million in 2020. | ||||||||||||
| Valuation Allowance | ||||||||||||||
| A valuation allowance has been established to offset the net deferred tax assets as realization of such assets is uncertain. We intend to continue maintaining a full valuation allowance on our DTAs until there is sufficient evidence to support the reversal of all or some portion of these allowances | We have considered future taxable income and tax planning strategies in assessing the need for a valuation allowance. Both future taxable income and tax planning strategies include a number of estimates. | Given our current earnings and anticipated future earnings, we believe that there is a reasonable possibility that within the next 12 months, sufficient positive evidence may become available to reach a conclusion that a significant portion of the valuation allowance will no longer be needed. Release of the valuation allowance would result in the recognition of certain DTAs and a decrease to income tax expense for the period the release is recorded. | ||||||||||||
Recent Accounting Pronouncements
Refer to Note 2, Summary of Significant Accounting Policies, of our consolidated financial statements for a discussion of recent accounting pronouncements and their effect, if any, on us.
43
Item 7A.Quantitative and Qualitative Disclosures About Market Risk
As of December 31, 2020, our cash equivalents and marketable securities consisted of investments in money market funds, asset-backed securities, U.S. Treasury securities, corporate debt obligations and commercial paper. These investments were made in accordance with our investment policy which specifies the categories, allocations, and ratings of securities we may consider for investment. The primary objective of our investment activities is to preserve principal while at the same time maximizing the income we receive without significantly increasing risk. Some of the financial instruments that we invest in could be subject to market risk. This means that a change in prevailing interest rates may cause the value of the instruments to fluctuate. For example, if we purchase a security that was issued with a fixed interest rate and the prevailing interest rate later rises, the value of that security will probably decline. Based on our current investment portfolio as of December 31, 2020, we do not believe that our results of operations would be materially impacted by an immediate change of 10% in interest rates.
We do not hold or issue derivatives, derivative commodity instruments or other financial instruments for speculative trading purposes. Further, we do not believe our cash, cash equivalents and marketable securities have significant risk of default or illiquidity. We made this determination based on discussions with our investment advisors and a review of our holdings. While we believe our cash, cash equivalents and marketable securities do not contain excessive risk, we cannot provide absolute assurance that in the future our investments will not be subject to adverse changes in market value. All of our cash equivalents and marketable securities are recorded at fair market value.
Item 8.Financial Statements and Supplementary Data
Our financial statements are annexed to this report beginning on page F-1.
Item 9.Changes In and Disagreements with Accountants on Accounting and Financial Disclosure
None.
44
Item 9A.Controls and Procedures
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our Exchange Act reports is recorded, processed, summarized and reported within the timelines specified in the Securities and Exchange Commission’s rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decision regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognized that any controls and procedures, no matter how well designed and operated, can only provide reasonable assurance of achieving the desired control objectives, and in reaching a reasonable level of assurance, management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
Under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, we conducted an evaluation of our disclosure controls and procedures, as such term is defined under Rule 13a-15(e) promulgated under the Securities Exchange Act of 1934, as amended. Based on this evaluation, our principal executive officer and our principal financial officer concluded that our disclosure controls and procedures were effective as of the end of the period covered by this Annual Report on Form 10-K.
Changes in Internal Control Over Financial Reporting
There have been no significant changes in our internal control over financial reporting that occurred during the quarter ended December 31, 2020 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) and Rule 15d-15(f) promulgated under the Exchange Act as a process designed by, or under the supervision of, our principal executive and principal financial officers and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
•Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets;
•Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures are being made only in accordance with authorizations of our management and directors; and
•Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on our financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2020. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control-Integrated Framework (2013 framework) (the COSO criteria). Based on our assessment, management concluded that, as of December 31, 2020, our internal control over financial reporting is effective based on the COSO criteria. The independent registered public accounting firm that audited the consolidated financial statements that are included in this Annual Report on Form 10-K has issued an audit report on the effectiveness of our internal control over financial reporting as of December 31, 2020. The report appears below.
45
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
Halozyme Therapeutics, Inc.
Opinion on Internal Control over Financial Reporting
We have audited Halozyme Therapeutics, Inc.’s internal control over financial reporting as of December 31, 2020, based on criteria established in Internal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). In our opinion, Halozyme Therapeutics, Inc. (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2020, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated balance sheets of the Company as of December 31, 2020 and 2019, and the related consolidated statements of operations, comprehensive (loss) income, cash flows, and stockholders’ equity (deficit) for each of the three years in the period ended December 31, 2020, and the related notes and the financial statement schedule listed in the Index at Item 15(a) and our report dated February 23, 2021 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
| /s/ Ernst & Young LLP | ||
San Diego, California
February 23, 2021
46
Item 9B.Other Information
None.
PART III
Item 10.Directors, Executive Officers and Corporate Governance
The information required by this item regarding directors is incorporated by reference to our definitive Proxy Statement (the Proxy Statement) to be filed with the Securities and Exchange Commission in connection with our 2021 Annual Meeting of Stockholders under the heading “Election of Directors.” The information required by this item regarding our code of ethics is incorporated by reference to the information under the caption “Code of Conduct and Ethics and Corporate Governance Guidelines” to be contained in the Proxy Statement. The information required by this item regarding our audit committee is incorporated by reference to the information under the caption “Board Meetings and Committees—Audit Committee” to be contained in the Proxy Statement. The information required by this item regarding material changes, if any, to the process by which stockholders may recommend nominees to our board of directors is incorporated by reference to the information under the caption “Board Meetings and Committees—Nominating and Governance Committee” to be contained in the Proxy Statement.
Executive Officers
Helen I. Torley, M.B. Ch. B., M.R.C.P. (58), President, Chief Executive Officer and Director. Dr. Torley joined Halozyme in January 2014 as President and Chief Executive Officer and as a member of Halozyme’s Board of Directors. Throughout her career, Dr. Torley has led several successful product launches, including Kyprolis®, Prolia®, Sensipar®, and Miacalcin®. Prior to joining Halozyme, Dr. Torley served as Executive Vice President and Chief Commercial Officer for Onyx Pharmaceuticals (Onyx) from August 2011 to December 2013 overseeing the collaboration with Bayer on Nexavar® and Stivarga® and the U.S. launch of Kyprolis. She was responsible for the development of Onyx's commercial capabilities in ex-US markets and in particular, in Europe. Prior to Onyx, Dr. Torley spent 10 years in management positions at Amgen Inc., most recently serving as Vice President and General Manager of the US Nephrology Business Unit from 2003 to 2009 and the U.S. Bone Health Business Unit from 2009 to 2011. From 1997 to 2002, she held various senior management positions at Bristol-Myers Squibb, including Regional Vice President of Cardiovascular and Metabolic Sales and Head of Cardiovascular Global Marketing. She began her career at Sandoz/Novartis, where she ultimately served as Vice President of Medical Affairs, developing and conducting post-marketing clinical studies across all therapeutic areas, including oncology. Dr. Torley serves on the board of directors of Quest Diagnostics Incorporated, a diagnostic information services company. Within the past five years, Dr. Torley served on the board of directors of Relypsa, Inc., a biopharmaceutical company. Before joining the industry, Dr. Torley was in medical practice as a senior registrar in rheumatology at the Royal Infirmary in Glasgow, Scotland. Dr. Torley received her Bachelor of Medicine and Bachelor of Surgery degrees (M.B. Ch.B.) from the University of Glasgow and is a Member of the Royal College of Physicians (M.R.C.P).
Elaine Sun (49), Senior Vice President, Chief Financial Officer. Ms. Sun joined Halozyme in March 2020 as Senior Vice President, Chief Financial Officer. Prior to joining Halozyme, from January 2017 to December 2019, Ms. Sun served in senior management positions at Sutrovax, Inc., a biopharmaceutical company, most recently serving as Chief Financial Officer and Chief Strategy Officer. From 2013 to December 2016, Ms. Sun was an independent financial advisory consultant for private and public healthcare companies. From 2009 to 2012, Ms. Sun served as Managing Director and Head of West Coast Healthcare at Evercore Partners, a leading M&A advisory firm, where she led Evercore’s U.S. life sciences efforts. From 2005 to 2008, Ms. Sun served as Managing Director, Healthcare Investment Banking at Merrill Lynch & Co. Ms. Sun received her MBA degree from Harvard Business School and her Bachelor of Arts degree from Wellesley College.
Masaru Matsuda (50), Senior Vice President, General Counsel, Chief Compliance Officer & Secretary. Mr. Matsuda joined Halozyme in August 2018 as Vice President, Associate General Counsel and Chief Compliance Officer and has served as Senior Vice President, General Counsel, Chief Compliance Officer and Secretary since January 2020. Prior to joining Halozyme, Mr. Matsuda held positions of increasing responsibility in the Law Department at Amgen Inc., a biopharmaceutical company, from May 2000 to August 2018, most recently as Vice President, Law, Global Commercial Operations. Prior to joining Amgen, Mr. Matsuda was an associate attorney at Orrick, Herrington & Sutcliffe LLP from June 1998 to April 2000,
47
and at Pillsbury Winthrop Shaw Pittman LLP from June 1996 to June 1998. Mr. Matsuda received his Juris Doctor from the University of California, Hastings College of the Law and his Bachelor of Science Degree in Business Administration from the University of Southern California.
Michael J. LaBarre (57), Senior Vice President, Chief Technical Officer. Dr. LaBarre joined Halozyme in June 2008 as Vice President, Product Development and has served in various officer positions most recently as Senior Vice President, Chief Technical Officer since January 2020. Prior to joining Halozyme, Dr. LaBarre served as Vice President, Product Development at Paramount BioSciences, a pharmaceutical company, from April 2006 to June 2008. Prior to that he served as Director, Analytical and Protein Biochemistry, Discovery Research at Biogen Idec, a pharmaceutical company, from December 2003 to April 2006. He also served in various research and development roles at IDEC Pharmaceuticals Corporation, a pharmaceutical company, from November 1995 to December 2003 most recently as Director, Analytical and Formulation Sciences, R&D. Prior to joining IDEC, Dr. LaBarre held research and development positions at various pharmaceutical companies from July 1992 to November 1995. Dr. LaBarre received his Ph.D. in Chemistry from the University of Arizona and his B.S. in Chemistry from Southampton College of Long Island University.
Item 11.Executive Compensation
The information required by this item is incorporated by reference to the information under the captions “Executive Compensation and Related Information” and “Compensation Committee Interlocks and Insider Participation” to be contained in the Proxy Statement.
Item 12.Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
Other than as set forth below, the information required by this item is incorporated by reference to the information under the caption “Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters” to be contained in the Proxy Statement.
Equity Compensation Plan Information
The following table summarizes our compensation plans under which our equity securities are authorized for issuance as of December 31, 2020:
| Plan Category | Number of Shares to be Issued upon Exercise of Outstanding Options, Restricted Stock Units and Performance Stock Units (a) | Weighted Average Exercise Price of Outstanding Options(2) (b) | Number of Shares Remaining Available for Future Issuance under Equity Compensation Plans (Excluding Shares Reflected in Column (a)) (c) | ||||||||||||||||||||
Equity compensation plans approved by stockholders (1) | 6,704,330 | $15.83 | 10,806,631 | ||||||||||||||||||||
| Equity compensation plans not approved by stockholders | — | — | — | ||||||||||||||||||||
| 6,704,330 | $15.83 | 10,806,631 | |||||||||||||||||||||
_____________________
(1)Represents stock options, restricted stock units, and performance stock units under the Amended and Restated 2011 Stock Plan.
(2)This amount does not include restricted stock units and performance stock units as there is no exercise price for such units.
48
Item 13.Certain Relationships and Related Transactions, and Director Independence
The information required by this item is incorporated by reference to the information under the caption “Certain Relationships and Related Transactions” and “Corporate Governance - Director Independence” to be contained in the Proxy Statement.
Item 14.Principal Accounting Fees and Services
The information required by this item is incorporated by reference to the information under the caption “Principal Accounting Fees and Services” to be contained in the Proxy Statement.
PART IV
Item 15.Exhibits and Financial Statement Schedules
(a)Documents filed as part of this report.
1. Financial Statements
| Page | ||||||||
| Report of Independent Registered Public Accounting Firm | F-1 | |||||||
| Consolidated Balance Sheets at December 31, 2020 and 2019 | F-3 | |||||||
| Consolidated Statements of Operations for Each of the Years Ended December 31, 2020, 2019 and 2018 | F-4 | |||||||
| Consolidated Statements of Comprehensive Income (Loss) for Each of the Years Ended December 31, 2020, 2019 and 2018 | F-5 | |||||||
| Consolidated Statements of Cash Flows for Each of the Years Ended December 31, 2020, 2019 and 2018 | F-6 | |||||||
| Consolidated Statements of Stockholders’ Equity for Each of the Years Ended December 31, 2020, 2019 and 2018 | F-8 | |||||||
| Notes to the Consolidated Financial Statements | F-9 | |||||||
2. List of all Financial Statement schedules.
The following financial statement schedule of Halozyme Therapeutics, Inc. is filed as part of this Annual Report on Form 10-K and should be read in conjunction with the consolidated financial statements of Halozyme Therapeutics, Inc.
| Page | ||||||||
| Schedule II: Valuation and Qualifying Accounts | F-43 | |||||||
All other schedules are omitted because they are not applicable or the required information is shown in the Financial Statements or notes thereto.
3. List of Exhibits required by Item 601 of Regulation S-K. See part (b) below.
49
(b)Exhibits.
| Incorporated by Reference | ||||||||||||||
| Exhibit | Filed | |||||||||||||
| Number | Exhibit Title | Herewith | Form | Date Filed | ||||||||||
| 3.1 | 8-K | 5/3/2019 | ||||||||||||
| 3.2 | 8-K | 12/19/2016 | ||||||||||||
| 4.1 | 8-K | 11/18/2019 | ||||||||||||
| 4.2 | 8-K | 11/18/2019 | ||||||||||||
| 4.3 | 10-K | 2/24/2020 | ||||||||||||
| 10.1 | SB-2 | 4/23/2004 | ||||||||||||
| 10.2 | 8-K | 1/12/2006 | ||||||||||||
| 10.3# | 8-K | 4/6/2018 | ||||||||||||
| 10.4# | 8-K | 5/6/2011 | ||||||||||||
| 10.5# | 8-K | 5/6/2011 | ||||||||||||
| 10.6# | 10-Q | 8/10/2015 | ||||||||||||
| 10.7# | 10-Q | 8/10/2015 | ||||||||||||
| 10.8# | 10-Q | 11/9/2015 | ||||||||||||
| 10.9# | 10-Q | 11/9/2015 | ||||||||||||
| 10.10# | 10-Q | 11/9/2015 | ||||||||||||
| 10.11# | 10-K | 2/28/2017 | ||||||||||||
| 10.12# | 8-K | 12/20/2007 | ||||||||||||
| 10.13# | 8-K | 12/13/2018 | ||||||||||||
| 10.14# | 10-Q | 11/9/2015 | ||||||||||||
| 10.15 | 8-K | 6/16/2011 | ||||||||||||
| 10.16 | 8-K | 7/5/2017 | ||||||||||||
50
| Incorporated by Reference | ||||||||||||||
| Exhibit | Filed | |||||||||||||
| Number | Exhibit Title | Herewith | Form | Date Filed | ||||||||||
| 10.17 | 10-Q | 5/10/2018 | ||||||||||||
| 10.18 | 8-K | 6/16/2011 | ||||||||||||
| 10.19 | 8-K | 7/5/2017 | ||||||||||||
| 10.20 | 10-Q | 5/10/2018 | ||||||||||||
| 10.21 | 10-K | 3/1/2013 | ||||||||||||
| 10.22 | 10-Q | 5/8/2013 | ||||||||||||
| 10.23 | 8-K | 7/5/2017 | ||||||||||||
| 10.24# | DEF-14A | 3/23/2016 | ||||||||||||
| 10.25# | 10-Q | 8/10/2020 | ||||||||||||
| 10.26# | X | |||||||||||||
| 21.1 | X | |||||||||||||
| 23.1 | X | |||||||||||||
| 31.1 | X | |||||||||||||
| 31.2 | X | |||||||||||||
| 32 | X | |||||||||||||
| 101.INS | XBRL Instance Document - the instance document does not appear in the interactive Data File because its XBRL tags are embedded within the Inline XBRL document. | X | ||||||||||||
| 101.SCH | XBRL Taxonomy Extension Schema | X | ||||||||||||
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase | X | ||||||||||||
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase | X | ||||||||||||
| 101.LAB | XBRL Taxonomy Extension Label Linkbase | X | ||||||||||||
| 101.PRE | XBRL Taxonomy Presentation Linkbase | X | ||||||||||||
| 104 | Cover Page Interactive Data File (formatted as inline XBRL and contained in Exhibit 101) | X | ||||||||||||
| # | Indicates management contract or compensatory plan or arrangement. | ||||
(c)Financial Statement Schedules. See Item 15(a) 2 above.
51
Item 16.Form 10-K Summary
None.
52
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| Halozyme Therapeutics, Inc., a Delaware corporation | ||||||||||||||||||||||||||
| Date: | February 23, 2021 | By: | /s/ Helen I. Torley, M.B. Ch.B., M.R.C.P. | |||||||||||||||||||||||
| Helen I. Torley, M.B. Ch.B., M.R.C.P. | ||||||||||||||||||||||||||
| President and Chief Executive Officer | ||||||||||||||||||||||||||
53
POWER OF ATTORNEY
Know all persons by these presents, that each person whose signature appears below constitutes and appoints Helen I. Torley and Elaine Sun, and each of them, as his/her true and lawful attorneys-in-fact and agents, with full power of substitution and resubstitution, for him/her and in his/her name, place, and stead, in any and all capacities, to sign any and all amendments to this Annual Report, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he/she might or could do in person, hereby ratifying and confirming that all said attorneys-in-fact and agents, or any of them or their or his/her substitute or substituted, may lawfully do or cause to be done by virtue thereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Annual Report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||||||||||||
| /s/ Helen I. Torley, M.B. Ch.B., M.R.C.P. | President and Chief Executive Officer | February 23, 2021 | ||||||||||||
| Helen I. Torley, M.B. Ch.B., M.R.C.P. | (Principal Executive Officer), Director | |||||||||||||
| /s/ Elaine Sun | Senior Vice President and Chief Financial Officer | February 23, 2021 | ||||||||||||
| Elaine Sun | (Principal Financial and Accounting Officer) | |||||||||||||
| /s/ Connie L. Matsui | Chair of the Board of Directors | February 23, 2021 | ||||||||||||
| Connie L. Matsui | ||||||||||||||
| /s/ Jean-Pierre Bizzari | Director | February 23, 2021 | ||||||||||||
| Jean-Pierre Bizzari | ||||||||||||||
| /s/ Bernadette Connaughton | Director | February 23, 2021 | ||||||||||||
| Bernadette Connaughton | ||||||||||||||
| /s/ James M. Daly | Director | February 23, 2021 | ||||||||||||
| James M. Daly | ||||||||||||||
| /s/ Jeffrey W. Henderson | Director | February 23, 2021 | ||||||||||||
| Jeffrey W. Henderson | ||||||||||||||
| /s/ Kenneth J. Kelley | Director | February 23, 2021 | ||||||||||||
| Kenneth J. Kelley | ||||||||||||||
| /s/ Matthew L. Posard | Director | February 23, 2021 | ||||||||||||
| Matthew L. Posard | ||||||||||||||
54
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
Halozyme Therapeutics, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Halozyme Therapeutics, Inc. (the Company) as of December 31, 2020 and 2019, and the related consolidated statements of operations, comprehensive (loss) income, stockholders’ equity (deficit) and cash flows for each of the three years in the period ended December 31, 2020, and the related notes and the financial statement schedule listed in the Index at Item 15(a) (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements referred to above, present fairly, in all material respects, the financial position of the Company at December 31, 2020 and 2019, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2020, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company’s internal control over financial reporting as of December 31, 2020, based on criteria established in Internal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated February 23, 2021 expressed an unqualified opinion thereon.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective or complex judgments. The communication of this critical audit matter does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing separate opinions on the critical audit matter or on the accounts or disclosures to which it relates.
F-1
| Estimation of Overall Transaction Price for Collaboration Agreements | ||||||||||||||
| Description of the Matter | At December 31, 2020 the Company has ten collaboration agreements. As discussed in Notes 2 and 4 of the financial statements, amounts are included in the transaction price when management determines that it is probable that the amount will not result in a significant reversal of revenue in the future. During 2020, the Company recognized $69.5 million of variable consideration in the transaction price under their collaboration arrangements. Auditing management’s conclusions related to determining the probability of achievement of milestones is complex and highly judgmental as a result of the uncertainties and limited visibility by the Company into the progression of developing and commercializing the combined targets as completed by the collaboration partners | |||||||||||||
| How We Addressed the Matter in Our Audit | We obtained an understanding and evaluated the design and tested the operating effectiveness of controls over the Company’s process to routinely evaluate the probability of achievement of milestones and any related constraint for each collaboration, in addition to the controls over the completeness and accuracy of determining the population of agreements and potential milestone payments. To test the milestone amounts included, or excluded, from the transaction price, we performed audit procedures that included, among others, observing the quarterly meetings with accounting and Alliance Managers discussing the status of each collaboration. For each milestone, we examined available evidence including correspondence with the collaboration partner and evaluated management’s conclusions on the probabilities of achievement. We reviewed supporting documentation to corroborate that milestones were included in the transaction price when determined to be probable of achievement. We reviewed the collaboration agreements and related amendments to validate the completeness of the list of targets and potential milestone payments that management considered in their analysis. We performed a lookback analysis to validate the company’s accuracy of determining the probability of achieving these milestones. | |||||||||||||
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 2006.
San Diego, California
February 23, 2021
F-2
HALOZYME THERAPEUTICS, INC.
CONSOLIDATED BALANCE SHEETS
(In thousands, except per share amounts)
| December 31, 2020 | December 31, 2019 | |||||||||||||
| ASSETS | ||||||||||||||
| Current assets: | ||||||||||||||
| Cash and cash equivalents | $ | 147,703 | $ | 120,179 | ||||||||||
| Marketable securities, available-for-sale | 220,310 | 301,083 | ||||||||||||
| Accounts receivable, net and other contract assets | 97,730 | 59,442 | ||||||||||||
| Inventories | 60,747 | 29,359 | ||||||||||||
| Prepaid expenses and other assets | 28,274 | 33,373 | ||||||||||||
| Total current assets | 554,764 | 543,436 | ||||||||||||
| Property and equipment, net | 10,593 | 10,855 | ||||||||||||
| Prepaid expenses and other assets | 14,067 | 11,083 | ||||||||||||
| Restricted cash | 500 | 500 | ||||||||||||
| Total assets | $ | 579,924 | $ | 565,874 | ||||||||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||||||||
| Current liabilities: | ||||||||||||||
| Accounts payable | $ | 1,928 | $ | 6,434 | ||||||||||
| Accrued expenses | 20,483 | 55,649 | ||||||||||||
| Deferred revenue, current portion | 1,746 | 4,012 | ||||||||||||
| Current portion of long-term debt, net | 397,228 | 19,542 | ||||||||||||
| Total current liabilities | 421,385 | 85,637 | ||||||||||||
| Deferred revenue, net of current portion | 4,026 | 1,247 | ||||||||||||
| Long-term debt, net | 0 | 383,045 | ||||||||||||
| Other long-term liabilities | 3,466 | 4,180 | ||||||||||||
| Commitments and contingencies (Note 9) | 0 | 0 | ||||||||||||
| Stockholders’ equity: | ||||||||||||||
| Preferred stock - $0.001 par value; 20,000 shares authorized; no shares issued and outstanding | 0 | 0 | ||||||||||||
| Common stock - $0.001 par value; 300,000 shares authorized; 135,030 and 136,713 shares issued and outstanding at December 31, 2020 and 2019, respectively | 135 | 137 | ||||||||||||
| Additional paid-in capital | 625,483 | 695,066 | ||||||||||||
| Accumulated other comprehensive loss | 22 | 240 | ||||||||||||
| Accumulated deficit | (474,593) | (603,678) | ||||||||||||
| Total stockholders’ equity | 151,047 | 91,765 | ||||||||||||
| Total liabilities and stockholders’ equity | $ | 579,924 | $ | 565,874 | ||||||||||
See accompanying notes to consolidated financial statements.
F-3
HALOZYME THERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands, except per share amounts)
| Year Ended December 31, | ||||||||||||||||||||
| 2020 | 2019 | 2018 | ||||||||||||||||||
| Revenues: | ||||||||||||||||||||
| Royalties | $ | 88,596 | $ | 69,899 | $ | 78,981 | ||||||||||||||
| Product sales, net | 55,987 | 66,048 | 28,234 | |||||||||||||||||
| Revenues under collaborative agreements | 123,011 | 60,045 | 44,647 | |||||||||||||||||
| Total revenues | 267,594 | 195,992 | 151,862 | |||||||||||||||||
| Operating expenses: | ||||||||||||||||||||
| Cost of product sales | 43,367 | 45,546 | 10,136 | |||||||||||||||||
| Research and development | 34,236 | 140,804 | 150,252 | |||||||||||||||||
| Selling, general and administrative | 45,736 | 77,252 | 60,804 | |||||||||||||||||
| Total operating expenses | 123,339 | 263,602 | 221,192 | |||||||||||||||||
| Operating income (loss) | 144,255 | (67,610) | (69,330) | |||||||||||||||||
| Other income (expense): | ||||||||||||||||||||
| Investment and other income, net | 5,425 | 6,986 | 7,578 | |||||||||||||||||
| Interest expense | (20,378) | (11,627) | (18,041) | |||||||||||||||||
| Net income (loss) before income taxes | 129,302 | (72,251) | (79,793) | |||||||||||||||||
| Income tax expense (benefit) | 217 | (11) | 537 | |||||||||||||||||
| Net income (loss) | $ | 129,085 | $ | (72,240) | $ | (80,330) | ||||||||||||||
| Net income (loss) per share: | ||||||||||||||||||||
| Basic | $ | 0.95 | $ | (0.50) | $ | (0.56) | ||||||||||||||
| Diluted | $ | 0.91 | $ | (0.50) | $ | (0.56) | ||||||||||||||
| Shares used in computing net income (loss) per share: | ||||||||||||||||||||
| Basic | 136,206 | 144,329 | 143,599 | |||||||||||||||||
| Diluted | 141,463 | 144,329 | 143,599 | |||||||||||||||||
See accompanying notes to consolidated financial statements.
F-4
HALOZYME THERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF COMPREHENSIVE INCOME (LOSS)
(In thousands)
| Year Ended December 31, | ||||||||||||||||||||
| 2020 | 2019 | 2018 | ||||||||||||||||||
| Net income (loss) | $ | 129,085 | $ | (72,240) | $ | (80,330) | ||||||||||||||
| Other comprehensive (loss) income: | ||||||||||||||||||||
| Unrealized (loss) gain on marketable securities | (164) | 508 | 182 | |||||||||||||||||
| Foreign currency translation adjustment | (32) | 9 | (8) | |||||||||||||||||
| Unrealized loss on foreign currency | (22) | 0 | (1) | |||||||||||||||||
| Total comprehensive income (loss) | $ | 128,867 | $ | (71,723) | $ | (80,157) | ||||||||||||||
See accompanying notes to consolidated financial statements.
F-5
HALOZYME THERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
| Year Ended December 31, | ||||||||||||||||||||
| 2020 | 2019 | 2018 | ||||||||||||||||||
| Operating activities: | ||||||||||||||||||||
| Net income (loss) | $ | 129,085 | $ | (72,240) | $ | (80,330) | ||||||||||||||
| Adjustments to reconcile net income (loss) to net cash provided by (used in) operating activities: | ||||||||||||||||||||
| Share-based compensation | 17,204 | 34,776 | 35,696 | |||||||||||||||||
| Depreciation and amortization | 3,284 | 4,068 | 2,388 | |||||||||||||||||
| Amortization of debt discount | 14,136 | 2,484 | 1,545 | |||||||||||||||||
| Amortization of premiums (accretion of discounts) on marketable securities | 839 | (2,469) | (3,090) | |||||||||||||||||
| (Gain) loss on disposal of equipment | (772) | 1,431 | 5 | |||||||||||||||||
| Deferral of unearned revenue | 4,632 | 0 | 3,000 | |||||||||||||||||
| Recognition of deferred revenue | (4,119) | (3,996) | (2,832) | |||||||||||||||||
| Deferral of lease payments | (1,033) | (459) | (7) | |||||||||||||||||
| Loss on impairment of right-of-use asset | 577 | 1,127 | 0 | |||||||||||||||||
| Loss on extinguishment of debt | 0 | 401 | 0 | |||||||||||||||||
| Other | (13) | (7) | (9) | |||||||||||||||||
| Changes in operating assets and liabilities: | ||||||||||||||||||||
| Accounts receivable, net | (38,288) | (29,437) | 11,613 | |||||||||||||||||
| Inventories | (31,388) | (6,734) | (17,480) | |||||||||||||||||
| Prepaid expenses and other assets | 2,518 | (19,006) | (5,695) | |||||||||||||||||
| Accounts payable and accrued expenses | (41,208) | 4,638 | 5,696 | |||||||||||||||||
| Net cash provided (used in) by operating activities | 55,454 | (85,423) | (49,500) | |||||||||||||||||
| Investing activities: | ||||||||||||||||||||
| Purchases of marketable securities | (226,185) | (389,759) | (311,112) | |||||||||||||||||
| Proceeds from maturities of marketable securities | 305,967 | 388,250 | 318,268 | |||||||||||||||||
| Purchases of property and equipment | (2,504) | (4,040) | (4,663) | |||||||||||||||||
| Proceeds from disposal of property and equipment | 1,076 | 0 | 0 | |||||||||||||||||
| Net cash provided by (used in) investing activities | 78,354 | (5,549) | 2,493 | |||||||||||||||||
| Financing activities: | ||||||||||||||||||||
| Proceeds from issuance of long-term debt, net | 0 | 447,350 | 0 | |||||||||||||||||
| Repayment of long-term debt | (19,560) | (108,082) | (77,516) | |||||||||||||||||
| Payment of debt issuance cost | 0 | (279) | 0 | |||||||||||||||||
| Repurchase of common stock | (150,117) | (199,998) | 0 | |||||||||||||||||
| Proceeds from issuance of common stock under equity incentive plans, net of taxes paid related to net share settlement | 63,393 | 14,224 | 13,719 | |||||||||||||||||
| Net cash (used in) provided by financing activities | (106,284) | 153,215 | $ | (63,797) | ||||||||||||||||
| Net increase (decrease) in cash, cash equivalents and restricted cash | 27,524 | 62,243 | (110,804) | |||||||||||||||||
| Cash, cash equivalents and restricted cash at beginning of period | 120,679 | 58,436 | 169,240 | |||||||||||||||||
| Cash, cash equivalents and restricted cash at end of period | $ | 148,203 | $ | 120,679 | $ | 58,436 | ||||||||||||||
F-6
| Year Ended December 31, | ||||||||||||||||||||
| 2020 | 2019 | 2018 | ||||||||||||||||||
| Supplemental disclosure of cash flow information: | ||||||||||||||||||||
| Interest paid | $ | 6,534 | $ | 9,029 | $ | 16,891 | ||||||||||||||
| Income taxes paid | $ | 180 | $ | 188 | $ | 220 | ||||||||||||||
| Supplemental disclosure of non-cash investing and financing activities: | ||||||||||||||||||||
| Amounts accrued for purchases of property and equipment | $ | 117 | $ | 61 | $ | 542 | ||||||||||||||
| Debt issuance cost included in accounts payable | $ | 0 | $ | 68 | $ | 0 | ||||||||||||||
| Right-of-use assets obtained in exchange for lease obligation | $ | 1,746 | $ | 897 | $ | 0 | ||||||||||||||
| Leasehold improvements paid by lessor | $ | 0 | $ | 0 | $ | 1,322 | ||||||||||||||
See accompanying notes to consolidated financial statements.
F-7
HALOZYME THERAPEUTICS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(in thousands)
| Common Stock | Additional Paid-In Capital | Accumulated Other Comprehensive Income (Loss) | Accumulated Deficit | Total Stockholders’ Equity (Deficit) | ||||||||||||||||||||||||||||||||||
| Shares | Amount | |||||||||||||||||||||||||||||||||||||
| BALANCE AT DECEMBER 31, 2017 | 142,789 | $ | 143 | $ | 731,044 | $ | (450) | $ | (522,371) | $ | 208,366 | |||||||||||||||||||||||||||
| Adjustment to beginning retained earnings | — | — | — | 71,263 | $ | 71,263 | ||||||||||||||||||||||||||||||||
| Share-based compensation expense | — | — | 35,696 | — | — | 35,696 | ||||||||||||||||||||||||||||||||
| Issuance of common stock pursuant to exercise of stock options and vesting of restricted stock units, net | 1,932 | 2 | 13,717 | — | — | 13,719 | ||||||||||||||||||||||||||||||||
| Issuance of restricted stock awards, net | 4 | 0 | 0 | — | — | 0 | ||||||||||||||||||||||||||||||||
| Other comprehensive income | — | — | — | 173 | — | 173 | ||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | (80,330) | (80,330) | ||||||||||||||||||||||||||||||||
| BALANCE AT DECEMBER 31, 2018 | 144,725 | $ | 145 | $ | 780,457 | $ | (277) | $ | (531,438) | $ | 248,887 | |||||||||||||||||||||||||||
| Share-based compensation expense | — | — | 34,776 | — | — | 34,776 | ||||||||||||||||||||||||||||||||
| Issuance of common stock pursuant to exercise of stock options and vesting of restricted stock units and performance restricted stock units, net | 2,493 | 2 | 14,222 | — | — | 14,224 | ||||||||||||||||||||||||||||||||
| Issuance of restricted stock awards, net | 74 | 0 | 0 | — | — | 0 | ||||||||||||||||||||||||||||||||
| Repurchase of common stock | (10,579) | (10) | (199,988) | (199,998) | ||||||||||||||||||||||||||||||||||
| Equity component of convertible notes | 65,599 | 65,599 | ||||||||||||||||||||||||||||||||||||
| Other comprehensive income | — | — | — | 517 | — | 517 | ||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | (72,240) | (72,240) | ||||||||||||||||||||||||||||||||
| BALANCE AT DECEMBER 31, 2019 | 136,713 | $ | 137 | $ | 695,066 | $ | 240 | $ | (603,678) | $ | 91,765 | |||||||||||||||||||||||||||
| Share-based compensation expense | — | — | 17,204 | — | — | 17,204 | ||||||||||||||||||||||||||||||||
| Issuance of common stock pursuant to exercise of stock options and vesting of restricted stock units and performance restricted stock units, net | 5,278 | 5 | 63,388 | — | — | 63,393 | ||||||||||||||||||||||||||||||||
| Issuance of restricted stock awards, net | 61 | 0 | 0 | — | — | 0 | ||||||||||||||||||||||||||||||||
| Repurchase of common stock | (7,022) | (7) | (150,110) | (150,117) | ||||||||||||||||||||||||||||||||||
| Equity component of convertible notes | (65) | (65) | ||||||||||||||||||||||||||||||||||||
| Other comprehensive loss | — | — | — | (218) | (218) | |||||||||||||||||||||||||||||||||
| Net income | — | — | — | — | 129,085 | 129,085 | ||||||||||||||||||||||||||||||||
| BALANCE AT DECEMBER 31, 2020 | 135,030 | $ | 135 | $ | 625,483 | $ | 22 | $ | (474,593) | $ | 151,047 | |||||||||||||||||||||||||||
See accompanying notes to consolidated financial statements.
F-8
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements
1. Organization and Business
Halozyme Therapeutics, Inc. is a biopharma technology platform company that provides innovative and disruptive solutions with the goal of improving patient experience and outcomes. Our proprietary enzyme, rHuPH20, is used to facilitate the delivery of injected drugs and fluids. We license our technology to biopharmaceutical companies to collaboratively develop products that combine our ENHANZE® drug delivery technology with the collaborators’ proprietary compounds.
Our approved product and our collaborators’ approved products and product candidates are based on rHuPH20, our patented recombinant human hyaluronidase enzyme. rHuPH20 is the active ingredient in our first commercially approved product, Hylenex® recombinant (“Hylenex”), and it works by breaking down hyaluronan (or “HA”), a naturally occurring carbohydrate that is a major component of the extracellular matrix in tissues throughout the body such as skin and cartilage. This temporarily increases dispersion and absorption allowing for improved subcutaneous delivery of injectable biologics, such as monoclonal antibodies and other large therapeutic molecules, as well as small molecules and fluids. We refer to the application of rHuPH20 to facilitate the delivery of other drugs or fluids as our ENHANZE® drug delivery technology (“ENHANZE”). We license the ENHANZE technology to form collaborations with biopharmaceutical companies that develop or market drugs requiring or benefiting from injection via the subcutaneous route of administration. In the development of proprietary intravenous (IV) drugs combined with our ENHANZE technology, data have been generated supporting the potential for ENHANZE to reduce treatment burden, as a result of shorter duration of subcutaneous (SC) administration. ENHANZE may enable fixed-dose SC dosing compared to weight-based dosing required for IV administration, and potentially allow for lower rates of infusion related reactions. ENHANZE may enable more flexible treatment options such as home administration by a healthcare professional or potentially the patient. Lastly, certain proprietary drugs co-formulated with ENHANZE have been granted additional exclusivity, extending the patent life of the product beyond the one of the proprietary IV drug.
We currently have ENHANZE collaborations with F. Hoffmann-La Roche, Ltd. and Hoffmann-La Roche, Inc. (“Roche”), Baxalta US Inc. and Baxalta GmbH (now members of the Takeda group of companies, following the acquisition of Shire plc by Takeda Pharmaceutical Company Limited in January 2019) (“Baxalta”), Pfizer Inc. (“Pfizer”), Janssen Biotech, Inc. (“Janssen”), AbbVie, Inc. (“AbbVie”), Eli Lilly and Company (“Lilly”), Bristol-Myers Squibb Company (“BMS”), Alexion Pharma Holding (“Alexion”), ARGENX BVBA (“argenx”) and Horizon Therapeutics plc. (Horizon). We receive royalties from three of these collaborations, including royalties from sales of one product from the Baxalta collaboration and three products from the Roche collaboration and one product from Janssen collaboration. Future potential revenues from royalties and fees from ENHANZE collaborations and the sales and/or royalties of our approved products will depend on the ability of Halozyme and our collaborators to develop, manufacture, secure and maintain regulatory approvals for approved products and product candidates and commercialize product candidates.
Except where specifically noted or the context otherwise requires, references to “Halozyme,” “the Company,” “we,” “our,” and “us” in these notes to consolidated financial statements refer to Halozyme Therapeutics, Inc. and its wholly owned subsidiary, Halozyme, Inc., and Halozyme, Inc.’s wholly owned subsidiaries, Halozyme Holdings Ltd., Halozyme Switzerland GmbH and Halozyme Switzerland Holdings GmbH.
F-9
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
2. Summary of Significant Accounting Policies
Basis of Presentation
The consolidated financial statements include the accounts of Halozyme Therapeutics, Inc. and our wholly owned subsidiary, Halozyme, Inc., and Halozyme, Inc.’s wholly owned subsidiaries, Halozyme Holdings Ltd., Halozyme Switzerland GmbH and Halozyme Switzerland Holdings GmbH. All intercompany accounts and transactions have been eliminated.
Use of Estimates
The preparation of consolidated financial statements in conformity with U.S. generally accepted accounting principles (“U.S. GAAP”) requires management to make estimates and assumptions that affect the amounts reported in our consolidated financial statements and accompanying notes. On an ongoing basis, we evaluate our estimates and judgments, which are based on historical and anticipated results and trends and on various other assumptions that management believes to be reasonable under the circumstances. By their nature, estimates are subject to an inherent degree of uncertainty and, as such, actual results may differ from management’s estimates.
Cash Equivalents and Marketable Securities
Cash equivalents consist of highly liquid investments, readily convertible to cash, that mature within ninety days or less from the date of purchase. As of December 31, 2020, our cash equivalents consisted of money market funds.
Marketable securities are investments with original maturities of more than ninety days from the date of purchase that are specifically identified to fund current operations. Marketable securities are considered available-for-sale. These investments are classified as current assets, even though the stated maturity date may be one year or more beyond the current balance sheet date which reflects management’s intention to use the proceeds from the sale of these investments to fund our operations, as necessary. Such available-for-sale investments are carried at fair value with unrealized gains and losses recorded in other comprehensive income (loss) and included as a separate component of stockholders’ equity. The cost of marketable securities is adjusted for amortization of premiums or accretion of discounts to maturity, and such amortization or accretion is included in investment and other income, net in the consolidated statements of operations. We use the specific identification method for calculating realized gains and losses on marketable securities sold. None of the realized gains and losses and declines in value judged to be as a result of credit loss on marketable securities, if any, are included in investment and other income, net in the consolidated statements of operations.
Restricted Cash
Under the terms of the leases of our facilities, we are required to maintain letters of credit as security deposits during the terms of such leases. At December 31, 2020 and 2019, restricted cash of $0.5 million was pledged as collateral for the letters of credit.
Fair Value of Financial Instruments
The authoritative guidance for fair value measurements establishes a three tier fair value hierarchy, which prioritizes the inputs used in measuring fair value. These tiers include: Level 1, defined as observable inputs such as quoted prices in active markets; Level 2, defined as inputs other than quoted prices in active markets that are either directly or indirectly observable; and Level 3, defined as unobservable inputs in which little or no market data exists, therefore requiring an entity to develop its own assumptions.
Our financial instruments include cash equivalents, available-for-sale marketable securities, accounts receivable, prepaid expenses and other assets, accounts payable, accrued expenses and short-term debt. Fair value estimates of these instruments are made at a specific point in time, based on relevant market information. These estimates may be subjective in nature and involve uncertainties and matters of significant judgment and therefore cannot be determined with precision. The carrying amount of cash equivalents, accounts receivable, prepaid expenses and other assets, accounts payable and accrued expenses are generally considered to be representative of their respective fair values because of the short-term nature of those instruments.
Available-for-sale marketable securities consist of asset-backed securities, corporate debt securities, U.S. Treasury securities and commercial paper, and are measured at fair value using Level 1 and Level 2 inputs. Level 2 financial instruments
F-10
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
are valued using market prices on less active markets and proprietary pricing valuation models with observable inputs, including interest rates, yield curves, maturity dates, issue dates, settlement dates, reported trades, broker-dealer quotes, issue spreads, benchmark securities or other market related data. We obtain the fair value of Level 2 investments from our investment manager, who obtains these fair values from a third-party pricing source. We validate the fair values of Level 2 financial instruments provided by our investment manager by comparing these fair values to a third-party pricing source.
Concentrations of Credit Risk, Sources of Supply and Significant Customers
We are subject to credit risk from our portfolio of cash equivalents and marketable securities. These investments were made in accordance with our investment policy which specifies the categories, allocations, and ratings of securities we may consider for investment. The primary objective of our investment activities is to preserve principal while at the same time maximizing the income we receive without significantly increasing risk. We maintain our cash and cash equivalent balances with one major commercial bank and marketable securities with another financial institution. Deposits held with the financial institutions exceed the amount of insurance provided on such deposits. We are exposed to credit risk in the event of a default by the financial institutions holding our cash, cash equivalents and marketable securities to the extent recorded on the consolidated balance sheets.
We are also subject to credit risk from our accounts receivable related to our product sales and revenues under our license and collaborative agreements. We have license and collaborative agreements with pharmaceutical companies under which we receive payments for royalties, license fees, milestone payments for specific achievements designated in the collaborative agreements, reimbursements of research and development services and supply of bulk formulation of rHuPH20. In addition, we sell Hylenex® recombinant in the United States to a limited number of established wholesale distributors in the pharmaceutical industry. Credit is extended based on an evaluation of the customer’s financial condition, and collateral is not required. Management monitors our exposure to accounts receivable by periodically evaluating the collectability of the accounts receivable based on a variety of factors including the length of time the receivables are past due, the financial health of the customer and historical experience. Based upon the review of these factors, we recorded 0 allowance for doubtful accounts at December 31, 2020 and 2019. Approximately 74% of the accounts receivable balance at December 31, 2020 represents amounts due from Janssen, Roche and Baxalta. Approximately 93% of the accounts receivable balance at December 31, 2019 represents amounts due from Janssen, Roche and Baxalta.
The following table indicates the percentage of total revenues in excess of 10% with any single customer:
| Year Ended December 31, | ||||||||||||||||||||
| 2020 | 2019 | 2018 | ||||||||||||||||||
| Partner A | 35% | 40% | 72% | |||||||||||||||||
| Partner B | 26% | 18% | 2% | |||||||||||||||||
| Partner C | 11% | 0% | 0% | |||||||||||||||||
| Partner D | 8% | 23% | 0% | |||||||||||||||||
F-11
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
We attribute revenues under collaborative agreements, including royalties, to the individual countries where the customer is headquartered. We attribute revenues from product sales to the individual countries to which the product is shipped. Worldwide revenues from external customers are summarized by geographic location in the following table (in thousands):
| Year Ended December 31, | ||||||||||||||||||||
| 2020 | 2019 | 2018 | ||||||||||||||||||
| United States | $ | 106,918 | $ | 28,178 | $ | 26,527 | ||||||||||||||
| Switzerland | 95,949 | 109,754 | 109,890 | |||||||||||||||||
| Ireland | 30,552 | 589 | 5,075 | |||||||||||||||||
| Belgium | 20,086 | 45,060 | 0 | |||||||||||||||||
| Japan | 10,644 | 9,905 | 8,873 | |||||||||||||||||
| All other foreign | 3,445 | 2,506 | 1,497 | |||||||||||||||||
| Total revenues | $ | 267,594 | $ | 195,992 | $ | 151,862 | ||||||||||||||
We rely on 2 third-party manufacturers for the supply of bulk rHuPH20 for use in the manufacture of Hylenex recombinant and our other collaboration products and product candidates. Payments due to these suppliers represented 75% and 47% of the accounts payable balance at December 31, 2020 and 2019, respectively. We also rely on a third-party manufacturer for the fill and finish of Hylenex recombinant product under a contract. Payments due to this supplier represented 0 and 8% of the accounts payable balance at December 31, 2020 and 2019, respectively.
Accounts Receivable, Net
Accounts receivable is recorded at the invoiced amount and is non-interest bearing. Accounts receivable is recorded net of allowances for doubtful accounts, cash discounts for prompt payment, distribution fees and chargebacks. We recorded 0 allowance for doubtful accounts at December 31, 2020 and 2019 as the collectability of accounts receivable was reasonably assured.
Inventories
Inventories are stated at lower of cost or net realizable value. Cost is determined on a first-in, first-out basis. Net realizable value is the estimated selling price in the ordinary course of business, less reasonably predictable costs of completion, disposal, and transportation. Inventories are reviewed periodically for potential excess, dated or obsolete status. We evaluate the carrying value of inventories on a regular basis, taking into account such factors as historical and anticipated future sales compared to quantities on hand, the price we expect to obtain for products in their respective markets compared with historical cost and the remaining shelf life of goods on hand.
As of December 31, 2020 and 2019, inventories consisted of $1.3 million and $1.4 million, respectively, of Hylenex inventory, net and $59.4 million and $28.0 million, respectively, of bulk rHuPH20, consistent with our plan to build inventory to meet future customer demand.
Leases
The Company has entered into operating leases primarily for real estate and automobiles. These leases have terms which range from 3 years to 6 years. We determine if an arrangement contains a lease at inception. Right of use (“ROU”) assets and liabilities resulting from operating leases are included in property and equipment, accrued expenses and other long-term liabilities on our consolidated balance sheets. Operating lease ROU assets and liabilities are recognized based on the present value of the future minimum lease payments over the lease term at commencement date. As most of our leases do not provide an implicit rate, we use our incremental borrowing rate based on the information available at commencement date in determining the discount rate to calculate the present value of future payments. The operating lease ROU asset also includes any lease payments made and excludes lease incentives and initial direct costs incurred. Our leases often include options to extend or terminate the lease. These options are included in the lease term when it is reasonably certain that we will exercise
F-12
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
that option. Short-term leases with an initial term of 12 months or less are not recorded on the balance sheet. Lease expense for minimum lease payments is recognized on a straight-line basis over the lease term.
We have lease agreements with lease and non-lease components, which are generally accounted for separately. For certain equipment leases, such as automobiles, we account for the lease and non-lease components as a single lease component.
Property and Equipment, Net
Property and equipment, including ROU assets are recorded at cost, less accumulated depreciation and amortization. Equipment is depreciated using the straight-line method over its estimated useful life ranging from three years to ten years and leasehold improvements are amortized using the straight-line method over the estimated useful life of the asset or the lease term, whichever is shorter.
Impairment of Long-Lived Assets
We account for long-lived assets in accordance with authoritative guidance for impairment or disposal of long-lived assets. Long-lived assets are reviewed for events or changes in circumstances, which indicate that their carrying value may not be recoverable.
Comprehensive Income (Loss)
Comprehensive income (loss) is defined as the change in equity during the period from transactions and other events and circumstances from non-owner sources.
Revenue Recognition
We generate revenues from payments received under collaborative agreements and product sales. We recognize revenue when we transfer promised goods or services to customers in an amount that reflects the consideration to which we expect to be entitled in exchange for those goods or services. To determine revenue recognition for contracts with customers we perform the following five steps: (i) identify the promised goods or services in the contract; (ii) identify the performance obligations in the contract, including whether they are distinct in the context of the contract; (iii) determine the transaction price, including the constraint on variable consideration; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) we satisfy the performance obligations.
Revenues under Collaborative Agreements
Under these agreements, we grant the collaboration partner a worldwide license to develop and commercialize products using our ENHANZE technology to combine our patented rHuPH20 enzyme with their proprietary biologics directed at up to a specified number of targets. Targets are usually licensed on an exclusive, global basis. Targets selected subsequent to inception of the arrangement require payment of an additional license fee. The collaboration partner is responsible for all development, manufacturing, clinical, regulatory, sales and marketing costs for any products developed under the agreement. We are responsible for supply of bulk rHuPH20 based on the collaboration partner’s purchase orders, and may also be separately engaged to perform research and development services. While these collaboration agreements are similar in that they originate from the same framework, each one is the result of an arms-length negotiation and thus may vary from one to the other.
We collect an upfront license payment from collaboration partners, and are also entitled to receive event-based payments subject to collaboration partners’ achievement of specified development, regulatory and sales-based milestones. In several agreements, collaboration partners pay us annual fees to maintain their exclusive license rights if they are unable to advance product development to specified stages. We earn separate fees for bulk rHuPH20 supplies and research and development services. In addition, collaboration partners will pay us royalties at an on average mid-single digit percent rate of their sales if products under the collaboration are commercialized. All amounts owed to us are noncancelable after the underlying triggering event occurs, and nonrefundable once paid. Unless terminated earlier in accordance with its terms, collaborations generally continue in effect until the last to expire royalty payment term, as determined on a product by product and on a country by country basis, with each royalty term starting on the first commercial sale of that product and ending the later of: (i) a specified period or term set forth in the agreement or (ii) expiration of the last to expire of the valid claims of our patents covering rHuPH20 or other specified patents developed under the collaboration which valid claim covers a product developed under the
F-13
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
collaboration. When there are no valid claims during the applicable royalty term in a given country, the royalty rate is reduced for those sales. Collaboration partners may terminate the agreement prior to expiration for any reason in its entirety or on a target-by-target basis generally upon 90 days prior written notice to us. Upon any such termination, the license granted to collaboration partners (in total or with respect to the terminated target, as applicable) will terminate provided, however, that in the event of expiration of the agreement (as opposed to a termination), the on-going licenses granted will become perpetual, non-exclusive and fully paid.
Although these agreements are in form identified as collaborative agreements, we concluded for accounting purposes they represent contracts with customers, and are not subject to accounting literature on collaborative arrangements. This is because we grant to collaboration partners licenses to our intellectual property, and provide supply of bulk rHuPH20 and research and development services which are all outputs of our ongoing activities, in exchange for consideration. Under these collaborative agreements, we do not develop assets jointly with collaboration partners, and do not share in significant risks of their development or commercialization activities. Accordingly, we concluded our collaborative agreements are appropriately accounted for pursuant to ASC Topic 606, Revenue from Contracts with Customers.
Under all of our collaborative agreements, we have identified licenses to use functional intellectual property as the only performance obligation. The intellectual property underlying the license is our proprietary ENHANZE® technology which represents application of rHuPH20 to facilitate delivery of drugs or fluids. Each of the licenses grants the collaboration partners rights to use our intellectual property as it exists and is identified on the effective date of the license, because there is no ongoing development of the ENHANZE technology required. Therefore, we recognize revenue from licenses at the point when the license becomes effective and the collaboration partner has received access to our intellectual property, usually at the inception of the agreement.
When collaboration partners can select additional targets to add to the licenses granted, we consider these rights to be options. We evaluate whether such options contain material rights, i.e. have exercise prices that are discounted compared to what we would charge for a similar license to a new collaboration partner. The exercise price of these options includes a combination of the target selection fees, event-based milestone payments and royalties. When these amounts in aggregate are not offered at a discount that exceeds discounts available to other customers, we conclude the option does not contain a material right, and we consider grants of additional licensing rights upon option exercises to be separate contracts (target selection contracts).
We provide customary indemnification and protection of licensed intellectual property for our customers. These provisions are part of assurance that the licenses meet the agreements’ representations and are not obligations to provide goods or services.
We also fulfill purchase orders for supply of bulk rHuPH20 and perform research and development services pursuant to projects authorization forms for our collaboration partners, which represent separate contracts. Additionally, we price our supply of bulk rHuPH20 and research and development services at our regular selling prices, called standalone selling price or SSP. Therefore, our collaboration partners do not have material rights to order these items at prices not reflective of SSP. Refer to the discussion below regarding recognition of revenue for these separate contracts.
Transaction price for a contract represents the amount to which we are entitled in exchange for providing goods and services to the customer. Transaction price does not include amounts subject to uncertainties unless it is probable that there will be no significant reversal of revenue when the uncertainty is resolved. Apart from the upfront license payment (or target selection fees in the target selection contracts), all other fees we may earn under our collaborative agreements are subject to significant uncertainties of product development. Achievement of many of the event-based development and regulatory milestones may not be probable until such milestones are actually achieved. This generally relates to milestones such as obtaining marketing authorization approvals. With respect to other development milestones, e.g. dosing of a first patient in a clinical trial, achievement could be considered probable prior to its actual occurrence, based on the progress towards commencement of the trial. In order to evaluate progress towards commencement of a trial, we assess the status of activities leading up to our collaboration partner’s initiation of a trial such as feedback received from the FDA (if applicable), completion of IND filings, readiness and availability of drug, readiness of study sites and our collaboration partner’s commitment of resources to the program. We do not include any amounts subject to uncertainties into the transaction price until it is probable that the amount will not result in a significant reversal of revenue in the future. At the end of each reporting period, we re-
F-14
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
evaluate the probability of achievement of such milestones and any related constraint, and if necessary, adjust our estimate of the overall transaction price.
When target exchange rights are held by collaboration partners, and the amounts attributed to these rights are not refundable, they are included in the transaction price. However, they are recorded as deferred revenues because we have a potential performance obligation to provide a new target upon an exchange right being exercised. These amounts are recognized in revenue when the right of exchange expires or is exercised.
Because our agreements have one type of performance obligation (licenses) which are typically all transferred at the same time at agreement inception, allocation of transaction price often is not required. However, allocation is required when licenses for some of the individual targets are subject to rights of exchange, because revenue associated with these targets cannot be recognized. We perform an allocation of the upfront amount based on relative SSP of licenses for individual targets. We determine license SSP using income-based valuation approach utilizing risk-adjusted discounted cash flow projections of the estimated return a licensor would receive. When amounts subject to uncertainties, such as milestones and royalties, are included in the transaction price, we attribute them to the specific individual target licenses which generate such milestone or royalty amounts.
We also estimate SSP of bulk rHuPH20 and research and development services, to determine that our collaboration partners do not have material rights to order them at discounted prices. For supplies of bulk rHuPH20, because we effectively act as a contract manufacturer to our collaboration partners, we estimate and charge SSP based on the typical contract manufacturer margins consistently with all of our collaborative partners. We determine SSP of research and development services based on a fully-burdened labor rate. Our rates are comparable to those we observe in other collaborative agreements. We also have a history of charging similar rates to all of our collaboration partners.
Upfront amounts allocated to licenses to individual targets are recognized as revenue when the license is transferred to the collaboration partner, as discussed above, if the license is not subject to exchange rights, or when the exchange right expires or is exercised. Development milestones and other fees are recognized in revenue when they are included in the transaction price, because by that time we have already transferred the related license to the collaboration partner.
Sales-based milestones and royalties cannot be recognized until the underlying sales occur. We do not receive final royalty reports from our collaboration partners until after we complete our financial statements for a prior quarter. Therefore, we recognize revenue based on estimates of the royalty earned, which are based on internal estimates and available preliminary reports provided by our collaboration partners. We will record a true-up in the following quarter if necessary, when final royalty reports are received. To date, we have not recorded any material true-ups.
In contracts to provide research and development services, such services represent the only performance obligation. The fees are charged based on hours worked by our employees and the fixed contractual rate per hour, plus third-party pass-through costs, on a monthly basis. We recognize revenues as the related services are performed based on the amounts billed, as the collaboration partner consumes the benefit of research and development work simultaneously as we perform these services, and the amounts billed reflect the value of these services to the customer.
Refer to Note 4 Revenue, for further discussion on our collaborative arrangements.
Product Sales, Net
Hylenex Recombinant
We sell Hylenex recombinant in the U.S. to wholesale pharmaceutical distributors, who sell the product to hospitals and other end-user customers. Sales to wholesalers are made pursuant to purchase orders subject to the terms of a master agreement, and delivery of individual packages of Hylenex recombinant represent performance obligations under each purchase order. We use a contract manufacturer to produce Hylenex recombinant and a third-party logistics (3PL) vendor to process and fulfill orders. We concluded we are the principal in the sales to wholesalers because we control access to services rendered by both vendors and direct their activities. We have no significant obligations to wholesalers to generate pull-through sales.
F-15
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
Selling prices initially billed to wholesalers are subject to discounts for prompt payment and subsequent chargebacks when wholesalers sell Hylenex recombinant at negotiated discounted prices to members of certain group purchasing organizations (“GPOs”) and government programs. We also pay quarterly distribution fees to certain wholesalers for inventory reporting and chargeback processing, and to GPOs as administrative fees for services and for access to GPO members. We concluded the benefits received in exchange for these fees are not distinct from our sales of Hylenex recombinant, and accordingly we apply these amounts to reduce revenues. Wholesalers also have rights to return unsold product nearing or past the expiration date. Because of the shelf life of Hylenex recombinant and our lengthy return period, there may be a significant period of time between when the product is shipped and when we issue credits on returned product.
We estimate the transaction price when we receive each purchase order taking into account the expected reductions of the selling price initially billed to the wholesaler arising from all of the above factors. We have compiled historical experience and data to estimate future returns and chargebacks of Hylenex recombinant and the impact of the other discounts and fees we pay. When estimating these adjustments to the transaction price, we reduce it sufficiently to be able to assert that it is probable that there will be no significant reversal of revenue when the ultimate adjustment amounts are known.
Each purchase order contains only one type of product, and is usually shipped to the wholesaler in a single shipment. Therefore, allocation of the transaction price to individual packages is not required.
We recognize revenue from Hylenex recombinant product sales and related cost of sales upon product delivery to the wholesaler location. At that time, the wholesalers take control of the product as they take title, bear the risk of loss of ownership, and have an enforceable obligation to pay us. They also have the ability to direct sales of product to their customers on terms and at prices they negotiate. Although wholesalers have product return rights, we do not believe they have a significant incentive to return the product to us.
Upon recognition of revenue from product sales of Hylenex recombinant, the estimated amounts of credit for product returns, chargebacks, distribution fees, prompt payment discounts, and GPO fees are included in sales reserves, accrued liabilities and net of accounts receivable. We monitor actual product returns, chargebacks, discounts and fees subsequent to the sale. If these amounts differ from our estimates, we make adjustments to these allowances, which are applied to increase or reduce product sales revenue and earnings in the period of adjustment.
In connection with the orders placed by wholesalers, we incur costs such as commissions to our sales representatives. However, as revenue from product sales is recognized upon delivery to the wholesaler, which occurs shortly after we receive a purchase order, we do not capitalize these commissions and other costs, based on application of the practical expedient allowed within the applicable guidance.
Bulk rHuPH20
We sell bulk rHuPH20 to collaboration partners for use in research and development; subsequent to receiving marketing approval, we sell it for use in collaboration commercial products. Sales are made pursuant to purchase orders subject to the terms of the collaborative agreement, and delivery of units of bulk rHuPH20 represent performance obligations under each purchase order. We provide a standard warranty that the product conforms to specifications. We use contract manufacturers to produce bulk rHuPH20 and have concluded we are the principal in the sales to collaboration partners. The transaction price for each purchase order of bulk rHuPH20 is fixed based on the cost of production plus a contractual markup, and is not subject to adjustments. Allocation of the transaction price to individual quantities of the product is usually not required because each order contains only one type of product.
We recognize revenue from the sale of bulk rHuPH20 as product sales and related cost of sales upon transfer of title to our partners. At that time, the partners take control of the product, bear the risk of loss of ownership, and have an enforceable obligation to pay us.
ENHANZE Drug Product
We sell ENHANZE drug product to collaboration partners for use in research and development in early phase clinical studies. Sales are made pursuant to purchase orders subject to the terms of the collaborative agreement, and delivery of units of ENHANZE drug product represent performance obligations under each purchase order. We provide a standard warranty that
F-16
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
the product conforms to specifications. We use contract manufacturers to produce ENHANZE drug product and we concluded we are the principal in the sales to collaboration partners. The transaction price for each purchase order of ENHANZE drug product is fixed based on the cost of production plus a contractual markup, and is not subject to adjustments. Allocation of the transaction price to individual quantities of the product is usually not required because each order contains only one type of product.
We recognize revenue from the sale of ENHANZE drug product as product sales and related cost of sales upon transfer of title to our partners. At that time, the partners take control of the product, bear the risk of loss of ownership, and have an enforceable obligation to pay us.
Revenue Presentation
In our statements of operations, we report as revenues under collaborative agreements the upfront payments, event-based development and regulatory milestones and sales milestones. We also include in this category revenues from separate research and development contracts pursuant to project authorization forms. We report royalties received from collaboration partners as a separate line in our statements of operations.
Revenues from sales of Hylenex recombinant, bulk rHuPH20 that has alternative future use and ENHANZE drug product are included in product sales, net.
In the footnotes to our financial statements, we provide disaggregated revenue information by type of arrangement (product sales, net, collaborative agreements and research and development services), and additionally, by type of payment stream received under collaborative agreements (upfront license fees, event-based development and regulatory milestones and other fees, sales milestones and royalties).
Cost of Product Sales
Cost of product sales consists primarily of raw materials, third-party manufacturing costs, fill and finish costs, freight costs, internal costs and manufacturing overhead associated with the production of Hylenex recombinant and bulk rHuPH20 and ENHANZE drug product. Cost of product sales also consists of the write-down of excess, dated and obsolete inventories and the write-off of inventories that do not meet certain product specifications, if any.
Research and Development Expenses
Research and development expenses include salaries and benefits, facilities and other overhead expenses, external clinical trial expenses, research related manufacturing services, contract services and other outside expenses. Research and development expenses are charged to operating expenses as incurred when these expenditures relate to our research and development efforts and have no alternative future uses. When bulk rHuPH20 is manufactured for use in research and development by us or our partners and the product cannot be redirected for alternative use due to formulation and manufacturing specifications, the manufacturing costs are recorded as research and development expense. Bulk rHuPH20 that is manufactured for partner use prior to our partner receiving marketing approval from the FDA or comparable regulatory agencies in foreign countries and meet these specifications is recorded as research and development expenses. Bulk rHuPH20 formulations manufactured for general partner and internal use, which can potentially be used by any collaboration partner or by us in Hylenex, is considered to have alternative future use and all manufacturing costs are capitalized as inventory.
We are obligated to make upfront payments upon execution of certain research and development agreements. Advance payments, including nonrefundable amounts, for goods or services that will be used or rendered for future research and development activities are deferred. Such amounts are recognized as expense as the related goods are delivered or the related services are performed or such time when we do not expect the goods to be delivered or services to be performed.
F-17
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
Milestone payments that we make in connection with in-licensed technology for a particular research and development project that have no alternative future uses (in other research and development projects or otherwise) and therefore no separate economic value are expensed as research and development costs at the time the costs are incurred. We currently have 0 in-licensed technologies that have alternative future uses in research and development projects or otherwise.
Clinical Trial Expenses
We make payments in connection with our clinical trials under contracts with contract research organizations that support conducting and managing clinical trials. The financial terms of these agreements are subject to negotiation and vary from contract to contract and may result in uneven payment flows. Generally, these agreements set forth the scope of work to be performed at a fixed fee, unit price or on a time and materials basis. A portion of our obligation to make payments under these contracts depends on factors such as the successful enrollment or treatment of patients or the completion of other clinical trial milestones.
Expenses related to clinical trials are accrued based on our estimates and/or representations from service providers regarding work performed, including actual level of patient enrollment, completion of patient studies and progress of the clinical trials. Other incidental costs related to patient enrollment or treatment are accrued when reasonably certain. If the amounts we are obligated to pay under our clinical trial agreements are modified (for instance, as a result of changes in the clinical trial protocol or scope of work to be performed), we adjust our accruals accordingly on a prospective basis. Revisions to our contractual payment obligations are charged to expense in the period in which the facts that give rise to the revision become reasonably certain.
Share-Based Compensation
We record compensation expense associated with stock options, restricted stock awards (“RSAs”), restricted stock units (“RSUs”) and performance stock units (“PSUs”) in accordance with the authoritative guidance for stock-based compensation. The cost of employee services received in exchange for an award of an equity instrument is measured at the grant date, based on the estimated fair value of the award, and is recognized as expense on a straight-line basis over the requisite service period of the award. Share-based compensation expense for an award with a performance condition is recognized when the achievement of such performance condition is determined to be probable. If the outcome of such performance condition is not determined to be probable or is not met, no compensation expense is recognized and any previously recognized compensation expense is reversed. Forfeitures are recognized as a reduction of share-based compensation expense as they occur.
Income Taxes
We provide for income taxes using the liability method. Under this method, deferred income tax assets and liabilities are determined based on the differences between the financial statement carrying amounts of existing assets and liabilities at each year end and their respective tax bases and are measured using enacted tax rates in effect for the year in which the differences are expected to affect taxable income. Significant judgment is required by management to determine our provision for income taxes, our deferred tax assets and liabilities, and the valuation allowance to record against our net deferred tax assets, which are based on complex and evolving tax regulations throughout the world. Deferred tax assets and other tax benefits are recorded when it is more likely than not that the position will be sustained upon audit. While we have begun to utilize certain of our net operating losses, we have not yet established a track record of profitability. Accordingly, valuation allowances have been recorded to reduce our net deferred tax assets to 0 until such time as we can demonstrate an ability to realize them.
F-18
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
In response to the coronavirus (COVID-19) pandemic, the Coronavirus Aid, Relief, and Economic Security Act (“CARES Act”) was enacted on March 27, 2020 in the U.S. The CARES Act includes many measures to assist companies, including temporary changes to income and non-income-based tax laws. One of the key tax provisions of the bill is allowing taxpayers with AMT credits to claim a refund in 2020 for the entire amount of the credit instead of recovering the credit through refunds over a period of years, as originally enacted by the Tax Cuts and Jobs Act (“TCJA”) in 2017. Under the TCJA, we had recorded a receivable for AMT credits that was expected to be received in future years. Under the CARES Act, the remaining receivable for the AMT credit is fully refundable in 2020. Other than the refundability of the AMT credit, at this time, we do not believe that the CARES Act will have a material impact on our financial statements. On December 27, 2020 the Consolidated Appropriations Act, 2021 was signed into law. It provides additional COVID-19 focused relief and extends certain provisions of the CARES Act. At this time, we do not believe that the Consolidated Appropriations Act, 2021 will have a material impact on our financial statements.
Net Income (Loss) Per Share
Basic net income (loss) per common share is computed by dividing net income (loss) for the period by the weighted average number of common shares outstanding during the period, without consideration for common stock equivalents. Outstanding stock options, unvested RSAs, unvested RSUs, unvested PSUs and the Convertible Notes are considered common stock equivalents and are only included in the calculation of diluted earnings per common share when net income is reported and their effect is dilutive. For the years ended December 31, 2020, 2019 and 2018, approximately 1.7 million, 33.1 million, and 13.8 million shares, respectively, of outstanding stock options, unvested RSAs, unvested RSUs, unvested PSUs and the Convertible Notes were excluded from the calculation of diluted net income (loss) per common share because their effect was anti-dilutive.
The 19.3 million shares underlying the conversion option of the Convertible Notes does not have an impact on our diluted earnings per share when the average market price of our common stock is less than the conversion price of $23.85 per share, as we will settle the principal amount of the Convertible Notes in cash upon conversion. When the average market price of our common stock exceeds the conversion price, we compute the potentially dilutive impact of the shares of common stock related to the Convertible Notes using the treasury stock method.
A reconciliation of the numerators and the denominators of the basic and diluted net income (loss) per common share computations is as follows (in thousands, except per share amounts):
| Year Ended December 31, | ||||||||||||||||||||
| 2020 | 2019 | 2018 | ||||||||||||||||||
| Numerator: | ||||||||||||||||||||
| Net income (loss) | $ | 129,085 | $ | (72,240) | $ | (80,330) | ||||||||||||||
| Denominator: | ||||||||||||||||||||
| Weighted average common shares outstanding for basic net income (loss) per share | 136,206 | 144,329 | 143,599 | |||||||||||||||||
| Net effect of dilutive common stock equivalents | 5,257 | 0 | 0 | |||||||||||||||||
| Weighted average common shares outstanding for diluted net income (loss) per share | 141,463 | 144,329 | 143,599 | |||||||||||||||||
| Net income (loss) per share: | ||||||||||||||||||||
| Basic | $ | 0.95 | $ | (0.50) | $ | (0.56) | ||||||||||||||
| Diluted | $ | 0.91 | $ | (0.50) | $ | (0.56) | ||||||||||||||
F-19
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
Segment Information
We operate our business in 1 segment, which includes all activities related to the research, development and commercialization of our proprietary enzymes. This segment also includes revenues and expenses related to (i) research and development and bulk rHuPH20 manufacturing activities conducted under our collaborative agreements with third parties and (ii) product sales of Hylenex recombinant. The chief operating decision-maker reviews the operating results on an aggregate basis and manages the operations as a single operating segment. Our long-lived assets located in foreign countries had no book value as of December 31, 2019 and 2018. There are no long-lived assets located in foreign countries as of December 31, 2020.
F-20
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
Adoption and Pending Adoption of Recent Accounting Pronouncements
The following table provides a brief description of recently issued accounting standards, those adopted in the current period and those not yet adopted:
| Standard | Description | Effective Date | Effect on the Financial Statements or Other Significant Matters | |||||||||||||||||
| In August 2020, the FASB issued ASU 2020-06, Debt with Conversion and other Options (Subtopic 470-20) and Derivatives and Hedging - Contracts in Entity’s Own Equity (Subtopic 815-40) | The new guidance eliminates the beneficial conversion and cash conversion accounting models for convertible instruments. It also amends the accounting for certain contracts in an entity’s own equity that are currently accounted for as derivatives because of specific settlement provisions. In addition, the new guidance requires that the if-converted method is used in computing diluted EPS for all convertible instruments | January 1, 2022 (Early adoption permitted effective January 1, 2021) | We plan to early adopt ASU 2020-06 as of January 1, 2021 on a modified retrospective basis, which is expected to result in an approximate $65.6 million decrease in additional paid in capital from the derecognition of the bifurcated equity component, $52.6 million increase in debt from the derecognition of the discount associated with the bifurcated equity component and $13.0 million decrease to the opening balance of accumulated deficit, representing the cumulative non-cash interest expense recognized related to the amortization of the bifurcated conversion option. We expect to write-off the related deferred tax liabilities of $11.8 million with a corresponding adjustment to the valuation allowance, resulting in no net impact to the cumulative adjustment to retained earnings. As we intended and have the ability to settle the principal amount of the convertible notes in cash upon conversion, in January 2021 we notified the note holders that we will settle the principal of the convertible notes in cash, removing our option to settle the principal of the notes in shares. Therefore, shares used for diluted EPS will continue to be limited to the excess conversion value over the principal amount of the convertible note. Diluted earnings per share will be impacted due to the elimination of non-cash interest expense associated with the amortization of the equity component. | |||||||||||||||||
| In August 2018, the FASB issued ASU 2018-15, Intangibles-Goodwill and other Internal-Use Software (Subtopic 350-40) | The new guidance aligns the requirement for capitalizing implementation costs incurred in a hosting arrangement that is a service contract with the requirement for capitalizing implementation costs incurred to develop or obtain internal-use software (and hosting arrangements that include an internal-use software license). | January 1, 2020 | We adopted the new guidance on January 1, 2020. The adoption did not have a material impact on our condensed consolidated financial position or results of operations. | |||||||||||||||||
F-21
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
| Standard | Description | Effective Date | Effect on the Financial Statements or Other Significant Matters | |||||||||||||||||
| In August 2018, the FASB issued ASU 2018-13, Fair Value Measurement (Topic 820). | The new guidance removes, modifies and adds to certain disclosure requirements on fair value measurements in Topic 820, Fair Value Measurement. | January 1, 2020 | We adopted the new guidance on January 1, 2020. The adoption did not have a material impact on our condensed consolidated financial position or results of operations. | |||||||||||||||||
| In June 2016, the FASB issued ASU 2016-13, Financial Instruments - Credit Losses (Topic 326), Measurement of Credit Losses on Financial Instruments | The standard amends the impairment model by requiring entities to use a forward-looking approach based on expected losses to estimate credit losses for most financial assets and certain other instruments that aren’t measured at fair value through net income. For available-for-sale debt securities, entities will be required to recognize an allowance for credit losses rather than a reduction in carrying value of the asset. Entities will no longer be permitted to consider the length of time that fair value has been less than amortized cost when evaluating when credit losses should be recognized. | January 1, 2020 | We adopted the new guidance on January 1, 2020. The adoption did not have a material impact on our condensed consolidated financial position or results of operations. | |||||||||||||||||
F-22
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
3. Fair Value Measurement
Available-for-sale marketable securities consisted of the following (in thousands):
| December 31, 2020 | ||||||||||||||||||||||||||
| Amortized Cost | Gross Unrealized Gains | Gross Unrealized Losses | Estimated Fair Value | |||||||||||||||||||||||
| Asset-backed securities | $ | 17,013 | $ | 49 | $ | 0 | $ | 17,062 | ||||||||||||||||||
| Corporate debt securities | 69,755 | 42 | (8) | 69,789 | ||||||||||||||||||||||
| U.S. Treasury securities | 45,110 | 7 | 0 | 45,117 | ||||||||||||||||||||||
| Commercial paper | 88,342 | 0 | 0 | 88,342 | ||||||||||||||||||||||
| $ | 220,220 | $ | 98 | $ | (8) | $ | 220,310 | |||||||||||||||||||
| December 31, 2019 | ||||||||||||||||||||||||||
| Amortized Cost | Gross Unrealized Gains | Gross Unrealized Losses | Estimated Fair Value | |||||||||||||||||||||||
| Asset-backed securities | $ | 30,484 | $ | 55 | $ | 0 | $ | 30,539 | ||||||||||||||||||
| Corporate debt securities | 161,308 | 178 | (14) | 161,472 | ||||||||||||||||||||||
| U.S. Treasury securities | 75,192 | 40 | (5) | 75,227 | ||||||||||||||||||||||
| Commercial paper | 33,845 | 0 | 0 | 33,845 | ||||||||||||||||||||||
| $ | 300,829 | $ | 273 | $ | (19) | $ | 301,083 | |||||||||||||||||||
As of December 31, 2020, 3 available-for-sale marketable securities with a fair market value of $21.2 million were in a gross unrealized loss position of $8 thousand. Based on our review of these marketable securities, we believe none of the unrealized loss is as a result of a credit loss as of December 31, 2020, because we do not intend to sell these securities and it is not more-likely-than-not that we will be required to sell these securities before the recovery of their amortized cost basis.
Contractual maturities of available-for-sale debt securities are as follows (in thousands):
| December 31, 2020 | December 31, 2019 | |||||||||||||
| Estimated Fair Value | ||||||||||||||
| Due within one year | $ | 220,310 | $ | 274,805 | ||||||||||
| After one but within five years | 0 | 26,278 | ||||||||||||
| $ | 220,310 | $ | 301,083 | |||||||||||
F-23
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
The following table summarizes, by major security type, our cash equivalents and available-for-sale marketable securities that are measured at fair value on a recurring basis and are categorized using the fair value hierarchy (in thousands):
| December 31, 2020 | December 31, 2019 | |||||||||||||||||||||||||||||||||||||
| Level 1 | Level 2 | Total estimated fair value | Level 1 | Level 2 | Total estimated fair value | |||||||||||||||||||||||||||||||||
| Cash equivalents: | ||||||||||||||||||||||||||||||||||||||
| Money market funds | $ | 140,571 | $ | 0 | $ | 140,571 | $ | 119,949 | $ | 0 | $ | 119,949 | ||||||||||||||||||||||||||
| Commercial paper | — | 7,000 | 7,000 | — | — | — | ||||||||||||||||||||||||||||||||
| Available-for-sale marketable securities: | ||||||||||||||||||||||||||||||||||||||
| Asset-backed securities | 0 | 17,062 | 17,062 | 0 | 30,539 | 30,539 | ||||||||||||||||||||||||||||||||
| Corporate debt securities | 0 | 69,789 | 69,789 | 0 | 161,472 | 161,472 | ||||||||||||||||||||||||||||||||
| U.S. Treasury securities | 45,117 | 0 | 45,117 | 75,227 | 0 | 75,227 | ||||||||||||||||||||||||||||||||
| Commercial paper | 0 | 88,342 | 88,342 | 0 | 33,845 | 33,845 | ||||||||||||||||||||||||||||||||
| $ | 185,688 | $ | 182,193 | $ | 367,881 | $ | 195,176 | $ | 225,856 | $ | 421,032 | |||||||||||||||||||||||||||
We had 0 instruments that were classified within Level 3 as of December 31, 2020 and 2019.
F-24
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
4. Revenue
Our disaggregated revenues were as follows (in thousands):
| Year Ended December 31, | ||||||||||||||||||||
| 2020 | 2019 | 2018 | ||||||||||||||||||
| Royalties | $ | 88,596 | $ | 69,899 | $ | 78,981 | ||||||||||||||
| Product sales, net | ||||||||||||||||||||
| Sales of bulk rHuPH20 | $ | 38,237 | $ | 48,285 | $ | 12,729 | ||||||||||||||
| Sales of ENHANZE drug product | 719 | 768 | 460 | |||||||||||||||||
| Sales of Hylenex | 17,031 | 16,995 | 15,045 | |||||||||||||||||
| Total product sales, net | 55,987 | 66,048 | 28,234 | |||||||||||||||||
| Revenues under collaborative agreements: | ||||||||||||||||||||
| Upfront license and target nomination fees | 37,264 | 53,000 | 26,336 | |||||||||||||||||
| Event-based development milestones and regulatory milestone and other fees | 69,500 | 5,500 | 16,000 | |||||||||||||||||
| Sales-based milestones | 15,000 | 0 | 0 | |||||||||||||||||
| Research and development services | 1,247 | 1,545 | 2,311 | |||||||||||||||||
| Total revenues under collaborative agreements | 123,011 | 60,045 | 44,647 | |||||||||||||||||
| Total revenue | $ | 267,594 | $ | 195,992 | $ | 151,862 | ||||||||||||||
During the year ended December 31, 2020 we recognized revenue related to licenses granted to collaboration partners in prior periods in the amount of $173.1 million. This amount represents royalties and sales milestones earned in the current period, as well as $69.5 million of variable consideration in the contracts where uncertainties have been resolved and the development milestones are probable of being achieved or were achieved. We also recognized revenue of $4.1 million during the year ended December 31, 2020 that had been included in deferred revenues at December 31, 2019. We did not recognize any adjustments to reduce sales reserves and allowances liability related to Hylenex recombinant sales in prior periods.
Upon the adoption of ASC 606, we recognized an adjustment to increase our accounts receivable by $19.4 million, decrease deferred revenues by $51.8 million, and decrease accumulated deficit by $71.2 million. The impact of applying the provisions of ASC 606 in the year ended December 31, 2018 was to decrease revenues by $4.7 million. Under the previously existing authoritative accounting literature, at December 31, 2018 our accounts receivable, net would have been $19.3 million lower, and our deferred revenue $47.4 million higher, than the amounts reported in our consolidated balance sheet. ASC 606 did not have an aggregate impact on our net cash used in operating activities, but resulted in offsetting changes in net loss and certain assets and liabilities within net cash used in operating activities in the consolidated statement of cash flows.
Accounts receivable, net, other contract assets and deferred revenues (contract liabilities) from contracts with customers, including collaboration partners, consisted of the following (in thousands):
| December 31, 2020 | December 31, 2019 | |||||||||||||
| Accounts receivable, net | $ | 90,730 | $ | 59,442 | ||||||||||
| Other contract assets | 7,000 | 0 | ||||||||||||
| Deferred revenues | 5,772 | 5,259 | ||||||||||||
As of December 31, 2020, the amounts included in the transaction price of our contracts with customers, including collaboration partners, and allocated to goods and services not yet provided were $55.3 million of which $49.5 million relates to
F-25
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
unfulfilled product purchase orders and $5.8 million has been collected and reported as deferred revenues. The unfulfilled product purchase orders are estimated to be delivered in 2021. Of the total deferred revenues of $5.8 million, $1.7 million is expected to be used by our customers within the next 12 months.
We recognized contract assets of $7.0 million at December 31, 2020, which relate to development milestones deemed probable of receipt for intellectual property licenses granted to collaboration partners in prior periods.
The following table presents amounts under our collaborative agreements included in the transaction price (i.e. cumulative amounts triggered or probable) as of December 31, 2020 (in thousands):
| Upfront (1) | Development (2) | Sales (3) | Total | |||||||||||||||||||||||
| Collaboration partner and agreement date: | ||||||||||||||||||||||||||
| Roche (December 2006, September 2017 and October 2018) | $ | 105,000 | $ | 47,000 | $ | 22,000 | $ | 174,000 | ||||||||||||||||||
| Baxalta (September 2007) | 10,000 | 3,000 | 9,000 | 22,000 | ||||||||||||||||||||||
| Pfizer (December 2012) | 14,500 | 2,000 | — | 16,500 | ||||||||||||||||||||||
| Janssen (December 2014) | 18,250 | 42,000 | 15,000 | 75,250 | ||||||||||||||||||||||
| AbbVie (June 2015) | 23,000 | 6,000 | — | 29,000 | ||||||||||||||||||||||
| Lilly (December 2015) | 33,000 | — | — | 33,000 | ||||||||||||||||||||||
| BMS (September 2017) | 110,000 | 10,000 | — | 120,000 | ||||||||||||||||||||||
| Alexion (December 2017) | 40,000 | 6,000 | — | 46,000 | ||||||||||||||||||||||
| argenx (February 2019) | 40,000 | 25,000 | 65,000 | |||||||||||||||||||||||
| Horizon (November 2020) | 30,000 | — | $ | — | 30,000 | |||||||||||||||||||||
| Royalties | 411,881 | |||||||||||||||||||||||||
| Total amounts under our collaborative agreements included in the transaction price | 1,022,631 | |||||||||||||||||||||||||
(1)Upfront and additional target selection fees
(2)Event-based development and regulatory milestone amounts and other fees
(3)Sales-based milestone amounts
Through December 31, 2020, our collaboration partners have completed development, obtained marketing authorization approvals for certain indications and commenced commercialization of the following products:
•Janssen, for DARZALEX FASPRO in US in May 2020 and subsequently in other regions.
•Roche, for Herceptin SC in the EU in August 2013 and subsequently in other regions; and MabThera SC in the EU in March 2014 and subsequently in other regions; and Phesgo in the US in June 2020 and subsequently in other regions.
•Baxalta, for HYQVIA in the EU and in the US in May 2013.
The remaining targets and products are currently in the process of development by the collaboration partners.
F-26
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
5. Certain Balance Sheet Items
Accounts receivable, net consisted of the following (in thousands):
| December 31, 2020 | December 31, 2019 | |||||||||||||
| Accounts receivable from product sales to collaborators | $ | 25,198 | $ | 35,649 | ||||||||||
| Accounts receivable from revenues under collaborative agreements | 30,404 | 3,850 | ||||||||||||
| Accounts receivable from royalty payments | 32,098 | 17,149 | ||||||||||||
| Accounts receivable from other product sales | 4,033 | 3,591 | ||||||||||||
| Other contract assets | 7,000 | 0 | ||||||||||||
| Subtotal | 98,733 | 60,239 | ||||||||||||
| Allowance for distribution fees and discounts | (1,003) | (797) | ||||||||||||
| Total accounts receivable, net | $ | 97,730 | $ | 59,442 | ||||||||||
Inventories consisted of the following (in thousands):
| December 31, 2020 | December 31, 2019 | |||||||||||||
| Raw materials | $ | 5,813 | $ | 2,769 | ||||||||||
| Work-in-process | 33,738 | 15,710 | ||||||||||||
| Finished goods | 21,196 | 10,880 | ||||||||||||
| Total inventories | $ | 60,747 | $ | 29,359 | ||||||||||
Prepaid expenses and other assets consisted of the following (in thousands):
| December 31, 2020 | December 31, 2019 | |||||||||||||
| Prepaid manufacturing expenses | $ | 35,048 | $ | 30,156 | ||||||||||
| Prepaid research and development expenses | 342 | 4,964 | ||||||||||||
| Other prepaid expenses | 2,510 | 3,655 | ||||||||||||
| Other assets | 4,441 | 5,681 | ||||||||||||
| Total prepaid expenses and other assets | 42,341 | 44,456 | ||||||||||||
| Less long-term portion | (14,067) | (11,083) | ||||||||||||
| Total prepaid expenses and other assets, current | $ | 28,274 | $ | 33,373 | ||||||||||
Prepaid manufacturing expenses include raw materials, slot reservation fees and other amounts paid to contract manufacturing organizations. Such amounts are reclassified to work-in-process inventory as materials are used or the CMO services are complete.
F-27
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
Property and equipment, net consisted of the following (in thousands):
| December 31, 2020 | December 31, 2019 | |||||||||||||
| Research equipment | $ | 7,085 | $ | 7,403 | ||||||||||
| Manufacturing equipment | 5,336 | 3,858 | ||||||||||||
| Computer and office equipment | 4,826 | 4,859 | ||||||||||||
| Leasehold improvements | 1,628 | 1,628 | ||||||||||||
| Subtotal | 18,875 | 17,748 | ||||||||||||
| Accumulated depreciation and amortization | (11,582) | (10,742) | ||||||||||||
| Subtotal | $ | 7,293 | $ | 7,006 | ||||||||||
| Right of use of assets | $ | 3,300 | $ | 3,849 | ||||||||||
| Property and equipment, net | $ | 10,593 | $ | 10,855 | ||||||||||
Depreciation and amortization expense was approximately $3.3 million, $4.1 million, and $2.4 million for the years ended December 31, 2020, 2019 and 2018, respectively. The depreciation and amortization expense is inclusive of $1.7 million, $1.8 million ROU asset amortization for the years ended December 31, 2020 and 2019, respectively.
Accrued expenses consisted of the following (in thousands):
| December 31, 2020 | December 31, 2019 | |||||||||||||
| Accrued outsourced research and development expenses | $ | 448 | $ | 8,423 | ||||||||||
| Accrued compensation and payroll taxes | 8,078 | 27,888 | ||||||||||||
| Accrued outsourced manufacturing expenses | 4,535 | 9,173 | ||||||||||||
| Other accrued expenses | 6,020 | 7,876 | ||||||||||||
| Lease liability | 4,868 | 6,469 | ||||||||||||
| Total accrued expenses | 23,949 | 59,829 | ||||||||||||
| Less long-term portion | (3,466) | (4,180) | ||||||||||||
| Total accrued expenses, current | $ | 20,483 | $ | 55,649 | ||||||||||
Expense associated with the accretion of the lease liabilities was approximately $0.5 million and $0.8 million for the twelve months ended December 31, 2020 and 2019, respectively. Total lease expense for the twelve months ended December 31, 2020 and 2019 $2.2 million and $2.6 million respectively.
Cash paid for amounts related to leases for the twelve months ended December 31, 2020 and 2019 was $3.2 million and $3.1 million respectively.
Deferred revenue consisted of the following (in thousands):
| December 31, 2020 | December 31, 2019 | |||||||||||||
| Collaborative agreements | ||||||||||||||
| License fees and event-based payments: | 0 | 2,764 | ||||||||||||
| Product sales | 5,772 | 2,495 | ||||||||||||
| Total deferred revenue | 5,772 | 5,259 | ||||||||||||
| Less current portion | (1,746) | (4,012) | ||||||||||||
| Deferred revenue, net of current portion | $ | 4,026 | $ | 1,247 | ||||||||||
F-28
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
6. Debt, Net
Convertible Notes
In November 2019, we completed the sale of $460.0 million in aggregate principal amount of 1.25% Convertible Senior Notes due 2024 (“Convertible Notes”) in a private placement to qualified institutional buyers pursuant to Rule 144A under the Securities Act of 1933, as amended (“Securities Act”). The Convertible Notes were issued under an indenture, dated as of November 18, 2019, (“Indenture”) with The Bank of New York Mellon Trust Company, N.A., as trustee. The offer and sale of the Convertible Notes and the shares of common stock issuable upon conversion of the Convertible Notes have not been registered under the Securities Act, or the securities laws of any other jurisdiction, and the Convertible Notes and such shares may not be offered or sold absent registration or an applicable exemption from registration requirements, or in a transaction not subject to, such registration requirements.
We received net proceeds from the offering of approximately $447.4 million. We used $200.0 million of the net proceeds from the offering to repurchase shares of common stock, including approximately $143.1 million to repurchase approximately 8.1 million shares of common stock concurrently with the offering in privately negotiated transactions, $6.9 million in open market purchases and $50.0 million to repurchase a total of approximately 2.6 million shares of common stock through an accelerated share repurchase agreement.
We used approximately $26.1 million of the net proceeds from the offering to repay all outstanding amounts under its loan agreement with Oxford Finance and Silicon Valley Bank and intend to use the remainder of the net proceeds for general corporate purposes, including additional share repurchases subsequent to the offering and working capital.
The Convertible Notes will pay interest semi-annually in arrears on June 1st and December 1st of each year, beginning on June 1, 2020, at an annual rate of 1.25%. As of December 31, 2020, the Convertible Notes were convertible into cash, shares of common stock or a combination of cash and shares of common stock, at our election, based on the applicable conversion rate at such time. The Convertible Notes are general unsecured obligations and will rank senior in right of payment to all indebtedness that is expressly subordinated in right of payment to the Convertible Notes, will rank equally in right of payment with all existing and future liabilities that are not so subordinated, will be effectively junior to any secured indebtedness to the extent of the value of the assets securing such indebtedness and will be structurally subordinated to all indebtedness and other liabilities (including trade payables) of the our current or future subsidiaries. The Convertible Notes have a maturity date of December 1, 2024.
Holders may convert their Convertible Notes at their option only in the following circumstances: (1) during any calendar quarter commencing after the calendar quarter ending on March 31, 2020, if the last reported sale price per share of common stock exceeds 130% of the conversion price for each of at least 20 trading days during the 30 consecutive trading days ending on, and including, the last trading day of the immediately preceding calendar quarter; (2) during the 5 consecutive business days immediately after any 5 consecutive trading day period (such 5 consecutive trading day period, the “measurement period”) in which the trading price per $1,000 principal amount of notes for each trading day of the measurement period was less than 98% of the product of the last reported sale price per share of Company’s common stock on such trading day and the conversion rate on such trading day; (3) upon the occurrence of certain corporate events or distributions on Company’s common stock, as described in the offering memorandum; (4) if we call such notes for redemption; and (5) at any time from, and including, June 1, 2024 until the close of business on the scheduled trading day immediately before the maturity date.
Upon the occurrence of certain circumstances, holders of the Convertible Notes may require us to purchase all or a portion of their notes for cash, which may require the use of a substantial amount of cash. As of December 31, 2020, the conditional conversion feature was triggered and our notes are classified as a current liability. We believe that it is remote that holders of the notes would choose to convert their notes early because the fair value of the security that a note holder can currently realize in an active market is greater than the conversion value the note holder would realize upon early conversion. For the year ended December 31, 2020, we have positive operating income and positive cash flow from operations and, accordingly, while there can be no assurance, we believe we have the ability to generate sufficient cash flows from operations or to raise additional capital through a variety of financing arrangements to satisfy early conversion of the Convertible Notes.
F-29
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
As a result of our plans to early adopt ASU 2020-06, in January 2021 we notified the note holders that we will settle the principal of the Convertible Notes in cash. Therefore, upon conversion, the principal value of the Convertible Notes will be paid in cash and depending on our stock price, any additional amount over principal amount will be settled in shares of common stock. The initial conversion rate for the Convertible Notes will be 41.9208 shares of common stock per $1,000 in principal amount of Convertible Notes, equivalent to a conversion price of approximately $23.85 per share of our common stock. The conversion rate is subject to adjustment as described in the Indenture.
In accordance with accounting guidance for debt with conversion and other options, we accounted for the debt and equity components of the Convertible Notes separately. The estimated fair value of the debt component at the date of issuance was $381.8 million, which was computed based on our non-convertible borrowing rate for similar debt of 5.19%, derived from independent valuation analysis. The equity component was allocated a value of $65.6 million and represents the difference between the $447.4 million of net proceeds from the issuance of the Convertible Notes and the $381.8 million estimated fair value of the debt component at the date of issuance.
In connection with the Convertible Notes, we paid the initial purchasers of the Convertible Notes a fee of $12.7 million and incurred additional debt issuance costs totaling $0.3 million, which includes expenses that we paid on behalf of the initial purchasers and expenses incurred directly by us. Debt issuance costs, the initial purchasers’ fee and the equity component is presented as a debt discount as of December 31, 2020 in the amount of $62.8 million, and will be amortized over the remaining estimated term of 3.9 using the effective interest method, utilizing an effective interest rate of 5.10%. The net carrying amount of the debt as of December 31, 2020 is $397.2 million. The fair value of the Convertible Notes, which was estimated using trading levels obtained from third-party service provider (Level 2), was $861.7 million at December 31, 2020 and $461.1 million at December 31, 2019.
For the year ended December 31, 2020 and 2019, we recognized interest expense of $19.9 million and $2.3 million including contractual coupon interest of $5.8 million and $0.7 million and amortization of the debt discount of $14.1 million and $1.6 million, respectively.
As of December 31, 2020, we were in compliance with all covenants under the Indenture and there was no material adverse change in our business, operations or financial condition.
Royalty-backed Loan
In January 2016, through our wholly-owned subsidiary Halozyme Royalty LLC (“Halozyme Royalty”), we received a $150 million loan (the “Royalty-backed Loan”) pursuant to a credit agreement (the “Credit Agreement”) with BioPharma Credit Investments IV Sub, LP and Athyrium Opportunities II Acquisition LP (the “Royalty-backed Lenders”). Under the terms of the Credit Agreement, Halozyme Therapeutics, Inc. transferred to Halozyme Royalty the right to receive royalty payments from the commercial sales of ENHANZE products owed under the Roche Collaboration and Baxalta Collaboration (“Collaboration Agreements”). The royalty payments from the Collaboration Agreements were used to repay the principal and interest on the loan (the “Royalty Payments”). The Royalty-backed Loan bore interest at a per annum rate of 8.75% plus the three-month LIBOR rate. The three-month LIBOR rate was subject to a floor of 0.7% and a cap of 1.5%. In June 2020, we paid the full remaining balance and final payment of $2.93 million thereby satisfying and discharging all obligations under, and terminating, the Royalty-backed Loan.
F-30
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
Oxford and SVB Loan and Security Agreement
In June 2016, we entered into a Loan and Security Agreement (the “Loan Agreement”) with Oxford Finance LLC (“Oxford”) and Silicon Valley Bank (“SVB”) (collectively, the “Lenders”), providing a senior secured loan facility of up to an aggregate principal amount of $70.0 million, comprising a $55.0 million draw in June 2016 and an additional $15.0 million tranche, which we had the option to draw during the second quarter of 2017 and did not exercise. The initial proceeds were partially used to pay the outstanding principal and final payment of $4.25 million owed on a previous loan agreement with the Lenders. The remaining proceeds were used for working capital and general business requirements. The senior secured loan facility carried a fixed interest rate of 8.25%. The repayment schedule provided for interest only payments for the first 18 months, followed by consecutive equal monthly payments of principal and interest in arrears through the maturity date of January 1, 2021. The Loan Agreement provided for a final payment equal to 5.50% of the initial $55.0 million principal amount, which was due when the Loan Agreement becomes due or upon the prepayment of the facility. We had the option to prepay the outstanding balance of the Loan Agreement in full and exercised this option in November 2019, at which point we paid the full remaining balance and final payment of $26.1 million, thereby satisfying and discharging all obligations under, and terminating, the Loan Agreement.
Future maturities and interest payments of long-term debt as of December 31, 2020, are as follows (in thousands):
| 2021 | $ | 465,750 | ||||||
| 2022 | 0 | |||||||
| 2023 | 0 | |||||||
| 2024 | 0 | |||||||
| 2025 | 0 | |||||||
| Total minimum payments | 465,750 | |||||||
| Less amount representing interest | (5,750) | |||||||
| Gross balance of long-term debt | 460,000 | |||||||
| Less unamortized debt discount | (62,772) | |||||||
| Present value of long-term debt | 397,228 | |||||||
| Less current portion of long-term debt | (397,228) | |||||||
| Long-term debt, less current portion and unamortized debt discount | $ | 0 | ||||||
F-31
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
7. Share-based Compensation
We currently grant stock options, restricted stock awards, performance stock units and restricted stock units under the Amended and Restated 2011 Stock Plan (“2011 Stock Plan”), which was approved by the stockholders on May 6, 2016 and provides for the grant of up to 44.2 million shares of common stock to selected employees, consultants and non-employee members of our Board of Directors as stock options, stock appreciation rights, restricted stock awards, restricted stock unit awards and performance awards. Awards are subject to terms and conditions established by the Compensation Committee of our Board of Directors. During the year ended December 31, 2020, we granted share-based awards under the 2011 Stock Plan. At December 31, 2020, 6,704,330 shares were subject to outstanding awards and 10,806,631 shares were available for future grants of share-based awards.
Total share-based compensation expense related to share-based awards was comprised of the following (in thousands):
| Year Ended December 31, | ||||||||||||||||||||
| 2020 | 2019 | 2018 | ||||||||||||||||||
| Research and development | $ | 5,484 | $ | 15,107 | $ | 17,220 | ||||||||||||||
| Selling, general and administrative | 11,720 | 19,669 | 18,476 | |||||||||||||||||
| Share-based compensation expense | $ | 17,204 | $ | 34,776 | $ | 35,696 | ||||||||||||||
Share-based compensation expense by type of share-based award (in thousands):
| Year Ended December 31, | ||||||||||||||||||||
| 2020 | 2019 | 2018 | ||||||||||||||||||
| Stock options | $ | 8,955 | $ | 17,624 | $ | 18,742 | ||||||||||||||
| RSAs, RSUs and PSUs | 8,249 | 17,152 | 16,954 | |||||||||||||||||
| $ | 17,204 | $ | 34,776 | $ | 35,696 | |||||||||||||||
Total unrecognized estimated compensation cost by type of award and the weighted-average remaining requisite service period over which such expense is expected to be recognized (in thousands, unless otherwise noted):
| December 31, 2020 | ||||||||||||||
| Unrecognized Expense | Remaining Weighted-Average Recognition Period (years) | |||||||||||||
| Stock options | $ | 18,853 | 2.60 | |||||||||||
| RSAs | $ | 460 | 0.33 | |||||||||||
| RSUs | $ | 13,241 | 2.32 | |||||||||||
| PSUs | $ | 297 | 1.75 | |||||||||||
Stock Options. Options granted under the Plans must have an exercise price equal to at least 100% of the fair market value of our common stock on the date of grant. The options generally have a maximum contractual term of ten years and vest at the rate of one-fourth of the shares on the first anniversary of the date of grant and 1/48 of the shares monthly thereafter. Certain option awards provide for accelerated vesting if there is a change in control (as defined in the Plans).
F-32
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
A summary of our stock option award activity as of and for the year ended December 31, 2020 is as follows:
| Shares Underlying Stock Options | Weighted Average Exercise Price per Share | Weighted Average Remaining Contractual Term (years) | Aggregate Intrinsic Value | |||||||||||||||||||||||
| Outstanding at December 31, 2019 | 11,548,229 | $14.72 | ||||||||||||||||||||||||
| Granted | 1,602,087 | $20.74 | ||||||||||||||||||||||||
| Exercised | (4,705,843) | $14.08 | ||||||||||||||||||||||||
| Canceled/forfeited | (2,810,851) | $17.02 | ||||||||||||||||||||||||
| Outstanding at December 31, 2020 | 5,633,622 | $15.83 | 6.58 | $151.4 | million | |||||||||||||||||||||
| Vested and expected to vest at December 31, 2020 | 5,633,622 | $15.83 | 6.58 | $151.4 | million | |||||||||||||||||||||
| Exercisable at December 31, 2020 | 3,297,904 | $13.36 | 5.08 | $96.8 | million | |||||||||||||||||||||
The weighted average grant date fair values of options granted during the years ended December 31, 2020, 2019 and 2018 were $20.74 per share, $16.46 per share and $10.33 per share, respectively. The total intrinsic value of options exercised during the years ended December 31, 2020, 2019 and 2018 was approximately $49.7 million, $10.6 million and $11.5 million, respectively. Cash received from stock option exercises for the years ended December 31, 2020, 2019 and 2018 was approximately $66.2 million, $16.5 million and $16.3 million, respectively.
The exercise price of stock options granted is equal to the closing price of the common stock on the date of grant. The fair value of each option award is estimated on the date of grant using the Black-Scholes-Merton option pricing model (“Black-Scholes model”). Expected volatility is based on historical volatility of our common stock. The expected term of options granted is based on analyses of historical employee termination rates and option exercises. The risk-free interest rate is based on the U.S. Treasury yield for a period consistent with the expected term of the option in effect at the time of the grant. The dividend yield assumption is based on the expectation of no future dividend payments. The assumptions used in the Black-Scholes model were as follows:
| Year Ended December 31, | ||||||||||||||||||||
| 2020 | 2019 | 2018 | ||||||||||||||||||
| Expected volatility | 47.57-51.82% | 51.56-56.94% | 57.18-70.06% | |||||||||||||||||
| Average expected term (in years) | 5.1 | 5.5 | 5.5 | |||||||||||||||||
| Risk-free interest rate | 0.22-1.67% | 1.35-2.56% | 2.25-2.96% | |||||||||||||||||
| Expected dividend yield | 0 | 0 | 0 | |||||||||||||||||
F-33
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
Restricted Stock Awards. RSAs are grants that entitle the holder to acquire shares of our common stock at 0 cost. The shares covered by a RSA cannot be sold, pledged, or otherwise disposed of until the award vests and any unvested shares may be reacquired by us for the original purchase price following the awardee’s termination of service. The RSAs will generally vest at the rate of one-fourth of the shares on each anniversary of the date of grant. Annual grants of RSAs to the Board of Directors typically vest in approximately one year.
The following table summarizes our RSA activity during the year ended December 31, 2020:
| Number of Shares | Weighted Average Grant Date Fair Value | |||||||||||||
| Unvested at December 31, 2019 | 211,123 | $11.47 | ||||||||||||
| Granted | 61,803 | $22.66 | ||||||||||||
| Vested | (210,676) | $11.48 | ||||||||||||
| Forfeited | (447) | $8.11 | ||||||||||||
| Unvested at December 31, 2020 | 61,803 | $22.66 | ||||||||||||
The estimated fair value of the RSAs was based on the closing market value of our common stock on the date of grant. The total grant date fair value of RSAs vested during the years ended December 31, 2020, 2019 and 2018 was approximately $2.4 million, $3.3 million and $4.5 million, respectively. The fair value of RSAs vested during the years ended December 31, 2020, 2019 and 2018, was approximately $4.3 million, $4.2 million and $7.2 million, respectively.
Restricted Stock Units. A RSU is a promise by us to issue a share of our common stock upon vesting of the unit. The RSUs will generally vest at the rate of one-fourth of the shares on each anniversary of the date of grant.
The following table summarizes our RSU activity during the year ended December 31, 2020:
| Number of Shares | Weighted Average Grant Date Fair Value | Weighted Average Remaining Contractual Term (yrs) | Aggregate Intrinsic Value | |||||||||||||||||||||||
| Outstanding at December 31, 2019 | 2,092,439 | $15.60 | ||||||||||||||||||||||||
| Granted | 574,279 | $20.25 | ||||||||||||||||||||||||
| Vested | (714,868) | $14.12 | ||||||||||||||||||||||||
| Forfeited | (921,938) | $16.62 | ||||||||||||||||||||||||
| Outstanding at December 31, 2020 | 1,029,912 | $18.31 | 1.23 | $44.0 | million | |||||||||||||||||||||
The estimated fair value of the RSUs was based on the closing market value of our common stock on the date of grant. The total grant date fair value of RSUs vested during the years ended December 31, 2020, 2019 and 2018 was approximately $10.1 million, $19.1 million and $6.7 million, respectively. The fair value of RSUs vested during the years ended December 31, 2020, 2019 and 2018 was approximately $14.0 million, $18.5 million and $11.0 million, respectively.
F-34
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
Performance Stock Units. A PSU is a promise by us to issue a share of our common stock upon achievement of a specific performance condition.
The following table summarizes our PSU activity during the year ended December 31, 2020:
| Number of Shares | Weighted Average Grant Date Fair Value | |||||||||||||
| Outstanding at December 31, 2019 | 0 | $0.00 | ||||||||||||
| Granted | 40,796 | $16.32 | ||||||||||||
| Vested | 0 | $0.00 | ||||||||||||
| Forfeited | 0 | $0.00 | ||||||||||||
| Outstanding at December 31, 2020 | 40,796 | $16.32 | ||||||||||||
The estimated fair value of the PSUs was based on the closing market value of our common stock on the date of grant. The fair value of PSUs vested during the years ended December 31, 2020, 2019 and 2018 was 0.
F-35
8. Stockholders’ Equity
During the years ended December 31, 2020, 2019 and 2018, we issued an aggregate of 4,705,843, 1,540,690 and 1,489,138 shares of common stock, respectively, in connection with the exercises of stock options, for net proceeds of approximately $66.2 million, $16.5 million and $16.3 million, respectively. For the years ended December 31, 2020, 2019 and 2018, we issued 571,963, 952,182 and 442,599 shares of common stock, respectively, upon vesting of certain RSUs for which the RSU holders surrendered 142,905, 140,466 and 139,850 RSUs, respectively, to pay for minimum withholding taxes totaling approximately $5.5 million, $7.0 million and $4.2 million, respectively. Stock options and unvested restricted units totaling approximately 6.7 million, 13.6 million and 13.4 million shares of our common stock were outstanding as of December 31, 2020, 2019 and 2018, respectively.
Share Repurchases
In November 2019, the Board of Directors authorized a capital return program to repurchase up to $550.0 million of outstanding common stock over a three-year period. We may utilize a variety of methods including open market purchases, privately negotiated transactions, accelerated share repurchase programs or any combination of such methods. The Board will regularly review this capital return program in connection with a balanced capital allocation strategy. During 2019, we repurchased approximately 11.1 million shares of common stock for $200.0 million at an average price of $18.03.
During 2020, we repurchased 6.5 million shares of common stock for $150.0 million at an average price of $23.05. The shares were purchased through open market transactions and through an ASR agreement with Bank of America in December 2020, for which we repurchased $21.7 million of common stock and received 0.5 million shares. We retired the repurchased shares and they resumed the status of authorized and unissued shares.
We had the following activity under the approved share repurchase programs (dollars in thousands, except share and per share data)
| 2020 | ||||||||||||||||||||
| Total Number of Shares Purchased | Weighted Average Price paid Per Share | Total Cost(2) | ||||||||||||||||||
First quarter(1) | 3,188,795 | $16.15 | $51,574 | |||||||||||||||||
| Second quarter | 88,307 | $22.58 | $1,996 | |||||||||||||||||
| Third quarter | 2,134,716 | $27.57 | $58,902 | |||||||||||||||||
Fourth quarter(3) | 1,095,366 | $34.36 | $37,645 | |||||||||||||||||
| 6,507,184 | $23.05 | $150,117 | ||||||||||||||||||
(1) This is in addition to 0.5 million shares delivered in February upon completion of the ASR.
(2) Included in the total cost of shares purchased is a commission fee of $0.02 per share.
(3) This includes the December 2020 ASR.
F-36
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
9. Commitments and Contingencies
Operating Leases
Our administrative offices and research facilities are located in San Diego, California. We lease an aggregate of approximately 50,000 square feet of office and research space in 2 buildings. The leases commenced in June 2011, November 2013 and June 2018 and continue through January 2023. The leases are subject to approximately 3.0% annual increases throughout the terms of the leases. We also pay a pro rata share of operating costs, insurance costs, utilities and real property taxes.
Additionally, we lease certain office equipment under operating leases. Total rent expense was approximately $2.3 million, $2.7 million and $2.5 million for the years ended December 31, 2020, 2019 and 2018, respectively.
Approximate annual future minimum operating lease payments as of December 31, 2020 are as follows (in thousands):
| Year: | Operating Leases | |||||||
| 2021 | $ | 2,563 | ||||||
| 2022 | 2,564 | |||||||
| 2023 | 150 | |||||||
| 2024 | 0 | |||||||
| 2025 | 0 | |||||||
| Total minimum lease payments | $ | 5,277 | ||||||
| Less imputed interest | (409) | |||||||
| Total | $ | 4,868 | ||||||
The weighted-average remaining lease term of our operating leases is approximately 2.06 years.
Other Commitments
We have existing supply agreements with contract manufacturing organizations Avid Bioservices, Inc. (“Avid”) and Catalent Indiana LLC (formerly Cook Pharmica LLC) (“Catalent”) to produce supplies of bulk rHuPH20. Under the terms of the agreements, we are committed to certain minimum annual purchases of bulk rHuPH20. At December 31, 2020, we had a $75.9 million minimum purchase obligation in connection with these agreements.
In June 2011, we entered into a services agreement with Patheon for the technology transfer and manufacture of Hylenex recombinant. At December 31, 2020, we had a $1.4 million minimum purchase obligation in connection with this agreement.
Legal Contingencies
From time to time, we may be involved in disputes, including litigation, relating to claims arising out of operations in the normal course of our business. Any of these claims could subject us to costly legal expenses and, while we generally believe that we have adequate insurance to cover many different types of liabilities, our insurance carriers may deny coverage or our policy limits may be inadequate to fully satisfy any damage awards or settlements. If this were to happen, the payment of any such awards could have a material adverse effect on our consolidated results of operations and financial position. Additionally, any such claims, whether or not successful, could damage our reputation and business. We currently are not a party to any legal proceedings, the adverse outcome of which, in management’s opinion, individually or in the aggregate, would have a material adverse effect on our consolidated results of operations or financial position.
F-37
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
10.Income Taxes
Total income (loss) before income taxes summarized by region were as follows (in thousands):
| Year Ended December 31, | ||||||||||||||||||||
| 2020 | 2019 | 2018 | ||||||||||||||||||
| United States | $ | 130,427 | $ | (70,737) | $ | (45,819) | ||||||||||||||
| Foreign | (1,125) | (1,514) | (33,974) | |||||||||||||||||
| Net income (loss) before income taxes | $ | 129,302 | $ | (72,251) | $ | (79,793) | ||||||||||||||
Significant components of our net deferred tax assets/(liabilities) were as follows (in thousands).
| December 31, | ||||||||||||||
| 2020 | 2019 | |||||||||||||
| Deferred tax assets: | ||||||||||||||
| Net operating loss carryforwards | $ | 84,278 | $ | 39,401 | ||||||||||
| Deferred revenue | 253 | 1,069 | ||||||||||||
| Research and development and orphan drug credits | 114,357 | 114,357 | ||||||||||||
| Share-based compensation | 4,637 | 9,972 | ||||||||||||
| Alternative minimum tax credit | 0 | 1,683 | ||||||||||||
| ASC 842 lease liability | 1,081 | 1,454 | ||||||||||||
| Interest expense limitation | 5,536 | 2,163 | ||||||||||||
| Other, net | 3,478 | 3,037 | ||||||||||||
| 213,620 | 173,136 | |||||||||||||
| Valuation allowance for deferred tax assets | (199,827) | (155,100) | ||||||||||||
| Deferred tax assets, net of valuation | 13,793 | 18,036 | ||||||||||||
| Deferred tax liabilities: | ||||||||||||||
| Depreciation | (1,002) | (865) | ||||||||||||
| Convertible note | (11,776) | (14,450) | ||||||||||||
| ASC 842 right of use asset | (733) | (865) | ||||||||||||
| Other, net | (282) | (173) | ||||||||||||
| Total deferred tax liabilities | (13,793) | (16,353) | ||||||||||||
| Net deferred tax asset | $ | 0 | $ | 1,683 | ||||||||||
A valuation allowance of $199.8 million and $155.1 million has been established to offset the net deferred tax assets as of December 31, 2020 and 2019, respectively, as realization of such assets is uncertain.
We intend to continue maintaining a full valuation allowance on our DTAs until there is sufficient evidence to support the reversal of all or some portion of these allowances. However, given our current earnings and anticipated future earnings, we believe that there is a reasonable possibility that within the next 12 months, sufficient positive evidence may become available to reach a conclusion that a significant portion of the valuation allowance will no longer be needed. Release of the valuation allowance would result in the recognition of certain DTAs and a decrease to income tax expense for the period the release is recorded. However, the exact timing and amount of the valuation allowance release are subject to change on the basis of the level of profitability that we are able to actually achieve.
F-38
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
Income tax expense was comprised of the following components (in thousands):
| Year Ended December 31, | ||||||||||||||||||||
| 2020 | 2019 | 2018 | ||||||||||||||||||
| Current - federal | $ | (11) | $ | 114 | $ | 82 | ||||||||||||||
| Current - state | 228 | (40) | 519 | |||||||||||||||||
| Deferred - federal | 0 | (85) | (64) | |||||||||||||||||
| Deferred - state | 0 | 0 | 0 | |||||||||||||||||
| $ | 217 | $ | (11) | $ | 537 | |||||||||||||||
The provision for income taxes on earnings subject to income taxes differs from the statutory federal income tax rate due to the following (in thousands):
| Year Ended December 31, | ||||||||||||||||||||
| 2020 | 2019 | 2018 | ||||||||||||||||||
| Federal income tax expense (benefit) at 21% | $ | 27,153 | $ | (15,173) | $ | (16,754) | ||||||||||||||
| State income tax benefit, net of federal income tax impact | (1,942) | (1,509) | (4,297) | |||||||||||||||||
| (Decrease) increase in valuation allowance | 44,727 | 8,147 | 35,731 | |||||||||||||||||
| Worthless stock deduction of international subsidiary | (67,322) | 0 | 0 | |||||||||||||||||
| Foreign income subject to tax at other than federal statutory rate | 237 | 318 | 7,106 | |||||||||||||||||
| Share-based compensation | (4,117) | 315 | (441) | |||||||||||||||||
| Executive compensation limitation | 1,434 | 858 | 866 | |||||||||||||||||
| Non-deductible expenses and other | 47 | 66 | 1,599 | |||||||||||||||||
| Research and development credits, net | 0 | (1,091) | (5,210) | |||||||||||||||||
| Orphan drug credits, net of federal add back | 0 | (5,718) | (18,063) | |||||||||||||||||
| Convertible note discount in APIC | $ | 0 | $ | 13,776 | $ | 0 | ||||||||||||||
| $ | 217 | $ | (11) | $ | 537 | |||||||||||||||
At December 31, 2020, our unrecognized tax benefit and uncertain tax positions were $19.2 million. Of this, $0.3 million of this amount would affect the effective tax rate and $18.9 million would affect the effective tax rate only in the event the valuation allowance was removed. Of the unrecognized tax benefits, we do not expect any significant changes to occur in the next 12 months. Interest and/or penalties related to uncertain income tax positions are recognized by us as a component of income tax expense. For the years ended December 31, 2020, 2019 and 2018, we recognized an immaterial amount of interest and penalties.
The following table summarizes the activity related to our unrecognized tax benefits (in thousands):
| Year Ended December 31, | ||||||||||||||||||||
| 2020 | 2019 | 2018 | ||||||||||||||||||
| Gross unrecognized tax benefits at beginning of period | $ | 21,483 | $ | 20,028 | $ | 14,428 | ||||||||||||||
| Increases in tax positions for prior years | 41 | 69 | 3,083 | |||||||||||||||||
| Decreases in tax positions for prior years | (2,357) | (23) | 0 | |||||||||||||||||
| Increases in tax positions for current year | 0 | 1,409 | 2,517 | |||||||||||||||||
| Gross unrecognized tax benefits at end of period | $ | 19,167 | $ | 21,483 | $ | 20,028 | ||||||||||||||
F-39
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
At December 31, 2020, we had federal, California and other state tax net operating loss carryforwards of approximately $310.8 million, $259.8 million and $45.3 million, respectively.
The following table shows key expiration dates of the federal and California net operating loss carryforwards (in thousands):
| Expires in: | ||||||||||||||||||||||||||
| Net Operating Loss | 2020 | 2021 and beyond | 2028 and beyond | |||||||||||||||||||||||
| Federal | $ | 310,756 | $ | 0 | $ | 310,756 | 0 | |||||||||||||||||||
| California | $ | 259,840 | $ | 0 | 0 | $ | 259,840 | |||||||||||||||||||
At December 31, 2020, we had federal and California research and development tax credit carryforwards of approximately $27.9 million and $19.1 million, respectively. The federal research and development tax credits will begin to expire in 2024 unless previously utilized. The California research and development tax credits will carryforward indefinitely until utilized. Additionally, we had Orphan Drug Credit carryforwards of $88.0 million which will begin to expire in 2034.
Pursuant to Internal Revenue Code Section 382, the annual use of the net operating loss carryforwards and research and development tax credits could be limited by any greater than 50% ownership change during any three year testing period. As a result of any such ownership change, portions of our net operating loss carryforwards and research and development tax credits are subject to annual limitations. We completed an updated Section 382 analysis regarding the limitation of the net operating losses and research and development credits as of December 31, 2019. Based upon the analysis, we determined that ownership changes occurred in prior years; however, the annual limitations on net operating loss and research and development tax credit carryforwards will not have a material impact on the future utilization of such carryforwards.
We do not provide for U.S. income taxes on the undistributed earnings of our foreign subsidiary as it is our intention to utilize those earnings in the foreign operations for an indefinite period of time. At December 31, 2020 and 2019, there were 0 undistributed earnings in foreign subsidiaries.
We are subject to taxation in the U.S. and in various state and foreign jurisdictions. Our tax years for 2004 and forward are subject to examination by the U.S. and California tax authorities due to the carryforward of unutilized net operating losses and research and development credits.
F-40
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
11.Employee Savings Plan
We have an employee savings plan pursuant to Section 401(k) of the Internal Revenue Code. All employees are eligible to participate, provided they meet the requirements of the plan. We are not required to make matching contributions under the plan. However, we voluntarily contributed to the plan approximately $1.1 million, $2.2 million and $1.3 million for the years ended December 31, 2020, 2019 and 2018, respectively.
F-41
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
12.Summary of Unaudited Quarterly Financial Information
The following is a summary of our unaudited quarterly results for the years ended December 31, 2020 and 2019 (in thousands):
| Quarter Ended | ||||||||||||||||||||||||||
| 2020 (Unaudited): | March 31, | June 30, | September 30, | December 31, | ||||||||||||||||||||||
Total revenues (1)(2)(3) | $ | 25,354 | $ | 55,221 | $ | 65,316 | $ | 121,703 | ||||||||||||||||||
| Gross profit on product sales | $ | 2,360 | $ | 597 | $ | 3,480 | $ | 6,183 | ||||||||||||||||||
| Total operating expenses | $ | 28,577 | $ | 25,666 | $ | 25,017 | $ | 44,079 | ||||||||||||||||||
| Net (loss) Income | $ | (6,103) | $ | 25,817 | $ | 36,207 | $ | 73,164 | ||||||||||||||||||
| Net (loss) Income per share: | ||||||||||||||||||||||||||
| Basic | $ | (0.04) | $ | 0.19 | $ | 0.27 | $ | 0.54 | ||||||||||||||||||
| Diluted | $ | (0.04) | $ | 0.19 | $ | 0.25 | $ | 0.50 | ||||||||||||||||||
| Shares used in computing net (loss) income per share: | ||||||||||||||||||||||||||
| Basic | 137,186 | 135,935 | 136,578 | 135,107 | ||||||||||||||||||||||
| Diluted | 137,186 | 138,084 | 142,081 | 145,122 | ||||||||||||||||||||||
| Quarter Ended | ||||||||||||||||||||||||||
| 2019 (Unaudited): | March 31, | June 30, | September 30, | December 31, | ||||||||||||||||||||||
Total revenues (4) | $ | 56,949 | $ | 39,148 | $ | 46,230 | $ | 53,665 | ||||||||||||||||||
| Gross profit on product sales | $ | 3,741 | $ | 3,883 | $ | 6,872 | $ | 6,006 | ||||||||||||||||||
Total operating expenses(5) | $ | 53,983 | $ | 53,125 | $ | 70,767 | $ | 85,727 | ||||||||||||||||||
| Net Income (loss) | $ | 1,796 | $ | (14,624) | $ | (25,015) | $ | (34,397) | ||||||||||||||||||
| Net Income (loss) per share: | ||||||||||||||||||||||||||
| Basic | $ | 0.01 | $ | (0.10) | $ | (0.17) | $ | (0.24) | ||||||||||||||||||
| Diluted | $ | 0.01 | $ | (0.10) | $ | (0.17) | $ | (0.24) | ||||||||||||||||||
| Shares used in computing net Income (loss) per share: | ||||||||||||||||||||||||||
| Basic | 144,743 | 145,411 | 146,136 | 141,046 | ||||||||||||||||||||||
| Diluted | 147,474 | 145,411 | 146,136 | 141,046 | ||||||||||||||||||||||
_______________
(1)Revenue for the quarter ended December 31, 2020 included $57.0 million in revenue under a collaborative agreement from Janssen, argenx, Horizon and BMS.
(2)Revenue for the quarter ended September 30, 2020 included $32.0 million in revenue under a collaborative arrangement from Roche and argenx.
(3)Revenue for the quarter ended June 30, 2020 included $32.3 million in revenue under a collaborative arrangement from Janssen and BMS.
(4)Revenue for the quarter ended March 31, 2019 included $30.0 million in revenue under a collaborative arrangement from argenx.
(5)Total operating expenses for the quarter ended December 31, 2019 included $28.4 million restructuring charges.
F-42
Halozyme Therapeutics, Inc.
Notes to Consolidated Financial Statements — (Continued)
Halozyme Therapeutics, Inc.
Schedule II
Valuation and Qualifying Accounts
(in thousands)
| Balance at Beginning of Period | Additions | Deductions | Balance at End of Period | |||||||||||||||||||||||
| For the year ended December 31, 2020 | ||||||||||||||||||||||||||
Accounts receivable allowances (1) | $ | 797 | $ | 13,276 | $ | (13,070) | $ | 1,003 | ||||||||||||||||||
| For the year ended December 31, 2019 | ||||||||||||||||||||||||||
Accounts receivable allowances (1) | $ | 592 | $ | 7,327 | $ | (7,122) | $ | 797 | ||||||||||||||||||
| For the year ended December 31, 2018 | ||||||||||||||||||||||||||
Accounts receivable allowances (1) | $ | 559 | $ | 5,988 | $ | (5,955) | $ | 592 | ||||||||||||||||||
_______________
(1)Allowances are for chargebacks, prompt payment discounts and distribution fees related to Hylenex recombinant product sales.
F-43