UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
| Form | 10-K | ||||
| ☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | ||||
For the fiscal year ended December 31, 2020
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | ||||
For the transition period from to
Commission File No. 001-36847

| Invitae Corporation | ||
| (Exact name of the registrant as specified in its charter) | ||
| Delaware | 27-1701898 | ||||
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) | ||||
1400 16th Street, San Francisco, California 94103
(Address of principal executive offices, Zip Code)
(415) 374-7782
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol | Name of exchange on which registered | ||||||||||||
| Common Stock, $0.0001 par value per share | NVTA | New York Stock Exchange | ||||||||||||
| Securities registered pursuant to Section 12(g) of the Act: None | ||
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☒ No ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☒ | Accelerated filer | ☐ | Non-accelerated filer | ☐ | Smaller reporting company | ☐ | Emerging growth company | ☐ | ||||||||||||||||||||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C.7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☒
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No ☒
As of June 30, 2020, the aggregate market value of common stock held by non-affiliates of the Registrant was approximately $4.0 billion, based on the closing price of the common stock as reported on The New York Stock Exchange for that date.
The number of shares of the registrant’s Common Stock outstanding as of February 19, 2021 was 196,654,925.
DOCUMENTS INCORPORATED BY REFERENCE
Items 10 (as to directors and Section 16(a) Beneficial Ownership Reporting Compliance), 11, 12, 13 and 14 of Part III incorporate by reference information from the registrant’s proxy statement to be filed with the Securities and Exchange Commission in connection with the solicitation of proxies for the registrant’s 2021 Annual Meeting of Stockholders.
TABLE OF CONTENTS
| Page No. | ||||||||
Forward-Looking Statements.
This report contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. All statements in this report other than statements of historical fact, including statements identified by words such as “believe,” “may,” “will,” “estimate,” “continue,” “anticipate,” “intend,” “expect” and similar expressions, are forward-looking statements. Forward-looking statements include, but are not limited to, statements about:
•our views regarding the future of genetic testing and its role in mainstream medical practice;
•the impact of COVID-19 on our business and the actions we may take in response thereto;
•our mission and strategy for our business, products and technology, including our ability to expand our content and develop new content while maintaining attractive pricing, further enhance our genetic testing service and the related user experience, build interest in and demand for our tests and attract potential partners;
•the implementation of our business model;
•the expected benefits from and our ability to integrate our acquisitions;
•the rate and degree of market acceptance of our tests and genetic testing generally;
•our ability to scale our infrastructure and operations in a cost-effective manner;
•the timing of and our ability to introduce improvements to our genetic testing platform and to expand our assays to include additional genes;
•the timing and results of studies with respect to our tests;
•developments and projections relating to our competitors and our industry;
•our competitive strengths;
•the degree to which individuals will share genetic information generally, as well as share any related potential economic opportunities with us;
•our commercial plans, including our sales and marketing expectations as well as our ability to expand internationally;
•our ability to obtain and maintain adequate reimbursement for our tests;
•regulatory developments in the United States and foreign countries;
•our ability to attract and retain key scientific or management personnel;
•our expectations regarding our ability to obtain and maintain intellectual property protection and not infringe on the rights of others;
•our ability to obtain funding for our operations and the growth of our business, including potential acquisitions;
•our financial performance;
•the impact of accounting pronouncements and our critical accounting policies, judgments, estimates and assumptions on our financial results;
•our expectations regarding our future revenue, cost of revenue, operating expenses and capital expenditures, and our future capital requirements; and
•the impact of tax laws on our business.
Forward-looking statements are subject to a number of risks and uncertainties that could cause actual results to differ materially from those expected. These risks and uncertainties include, but are not limited to, those risks discussed in Item 1A of this report. Although we believe that the expectations and assumptions reflected in the forward-looking statements are reasonable, we cannot guarantee future results, level of activity, performance or achievements. In addition, neither we nor any other person assumes responsibility for the accuracy and completeness of any of these forward-looking statements. Any forward-looking statements in this report speak only as of the date of this report. We expressly disclaim any obligation or undertaking to update any forward-looking statements.
This report contains statistical data and estimates that we obtained from industry publications and reports. These publications typically indicate that they have obtained their information from sources they believe to be reliable, but do not guarantee the accuracy and completeness of their information. Some data contained in this report is also based on our internal estimates. Although we have not independently verified the third-party data, we believe it to be reasonable.
In this report, all references to “Invitae,” “we,” “us,” “our,” or “the Company” mean Invitae Corporation.
Invitae and the Invitae logo are trademarks of Invitae Corporation. We also refer to trademarks of other companies and organizations in this report.
1
Summary of Risk Factors.
Our business is subject to numerous risks and uncertainties that could affect our ability to successfully implement our business strategy and affect our financial results. You should carefully consider all of the information in this report and, in particular, the following principal risks and all of the other specific factors described in Item 1A. of this report, “Risk Factors,” before deciding whether to invest in our company.
•We face risks related to health epidemics, including the recent COVID-19 pandemic, which could have a material adverse effect on our business and results of operations.
•We expect to continue incurring significant losses, and we may not successfully execute our plan to achieve or sustain profitability.
•Our inability to raise additional capital on acceptable terms in the future may limit our ability to develop and commercialize new tests and expand our operations.
•We have acquired and may continue to acquire businesses or assets, form joint ventures or make investments in other companies or technologies that could harm our operating results, dilute our stockholders’ ownership, or cause us to incur debt or significant expense.
•If third-party payers, including managed care organizations, private health insurers and government health plans do not provide adequate reimbursement for our tests or we are unable to comply with their requirements for reimbursement, our commercial success could be negatively affected.
•We face intense competition, which is likely to intensify further as existing competitors devote additional resources to, and new participants enter, the market. If we cannot compete successfully, we may be unable to increase our revenue or achieve and sustain profitability.
•We may not be able to manage our future growth effectively, which could make it difficult to execute our business strategy.
•Security breaches, loss of data and other disruptions could compromise sensitive information related to our business or prevent us from accessing critical information and expose us to liability, which could adversely affect our business and our reputation.
•Litigation or other proceedings or third-party claims of intellectual property infringement or misappropriation, including ongoing litigation with respect to alleged intellectual property infringement against ArcherDX, Inc., or ArcherDX, will require us to spend significant time and money, could, in the future, prevent us from selling certain of our tests, and could have a material adverse effect on our business, financial condition and stock price.
•We rely on highly skilled personnel in a broad array of disciplines and, if we are unable to hire, retain or motivate these individuals, or maintain our corporate culture, we may not be able to maintain the quality of our services or grow effectively.
•We need to scale our infrastructure in advance of demand for our tests, and our failure to generate sufficient demand for our tests would have a negative impact on our business and our ability to attain profitability.
•If we are not able to continue to generate substantial demand of our tests, our commercial success will be negatively affected.
•If ArcherDX’s products and services do not perform as expected, we may not realize the expected benefits of our recent acquisition of ArcherDX.
•The future growth of our distributed products business is partially dependent upon regulatory approval and market acceptance of our in vitro diagnostic, or IVD products, including STRATAFIDE and Personalized Cancer Monitoring, or PCM.
•Our success will depend on our ability to use rapidly changing genetic data to interpret test results accurately and consistently, and our failure to do so would have an adverse effect on our operating results and business, harm our reputation and could result in substantial liabilities that exceed our resources.
2
PART I
ITEM 1. Business
Overview
Invitae is in the business of delivering genetic testing services that support a lifetime of patient care – from inherited disease diagnoses, to family planning, to proactive health screening to personalized diagnosis, treatment and monitoring of cancer. Those tests are delivered via a unique, rapidly expanding platform that serves patients, healthcare providers, biopharma companies and other partners, thereby capturing the broad potential of genetics and helping to expand its use across the healthcare continuum. Invitae applies proprietary design, process automation, robotics and bioinformatics software solutions to achieve efficiencies in sample processing and complex variant interpretation, allowing medical interpretation at scale. The result is a new and simplified process for obtaining and using affordable, high-quality genetic information to inform critical healthcare decisions. That access and scale also enable genomic information to speed the discovery and development of new personalized medical therapies — all while making clinical genetic testing available to billions of people.
By pioneering new ways of sharing, understanding and applying genetic information, Invitae is transforming the field of genetics from a series of one-time, one-dimensional queries to a lifelong clinical dialogue with our genes using complex analyses and information management to improve medical decisions and optimize health interventions.
Mission and strategy
Invitae’s mission is to bring comprehensive genetic information into mainstream medical practice to improve the quality of healthcare for billions of people. Our goal is to aggregate a majority of the world’s genetic information into a comprehensive network that enables sharing of data among network participants to improve healthcare and clinical outcomes.
We were founded on four core principles:
•Patients should own and control their own genetic information;
•Healthcare professionals are fundamental in ordering and interpreting genetic information;
•Driving down the price of genetic information will increase its clinical and personal utility; and
•Genetic information is more valuable when shared.
Our strategy for long-term growth centers on five key drivers of our business, which we believe work in conjunction to create a flywheel effect extending our leadership position in the new market we are building:

•Expanding our content offering. We intend to continue steadily adding additional testing and analysis content to the Invitae platform, ultimately leading to affordable and ongoing access to the molecular information that enables personalized medicine. The breadth and depth of our offering is a core and central contribution to an improved user experience.
3
•Creating a unique user experience. A state-of-the-art interactive platform will enhance our service offering, leverage the uniquely empowering characteristics of online sharing of genetic information and, we believe, enable a superior economic offering to clients. We intend to continue to expend substantial efforts developing, acquiring and implementing technology-driven improvements to our customers’ experience. We believe that an enhanced user experience and the resulting benefits to our brand and reputation will help draw customers to us over and above our direct efforts to do so.
•Driving volume. We intend to increase our brand equity and visibility through a commitment to precision testing results, excellent service and a variety of marketing and promotional techniques, including scientific publications and presentations, sales, marketing, public relations, social media and web technology vehicles. We believe that rapidly increasing the number of customers using our platform helps us to attract partners.
•Attracting partners. As we add more customers to our platform, we believe our business becomes particularly attractive to potential partners that can help the patients in our network further benefit from their genetic information or that provide us access to new customers who may wish to join our network. We believe the cumulative effect of the increased volume brought by these strategic components will allow us to lower the cost of our service and expand patient access globally.
•Lowering the cost and price of genetic information. Our goal is to provide customers with a broad menu of genetic content at a reasonable price and rapid turn-around times in order to grow volume and, in turn, achieve greater economies of scale. As our customers and our business benefit from further cost savings, we expect that those cost savings will allow us to deliver still more comprehensive information at decreasing prices and further improve the customer experience, allowing us to reap the cumulative benefits from all of the efforts outlined above.
We seek to differentiate our service in the market by establishing an exceptional experience for our customers. To that end, we believe that elevating the needs of the customer over those of our other stakeholders is essential to our success. Thus, in our decision-making processes, we strive to prioritize, in order:
•The needs of our customers;
•Motivating our employees to serve our customers; and
•Our long-term stockholder value.
We are certain that focusing on customers as our top priority rather than short-term financial goals is the best way to build and operate an organization for maximum long-term value creation.
Business overview
We are focused on making comprehensive, high-quality genetic information more accessible by lowering the cost of genetic testing, by utilizing a testing delivery platform that is accessible to patients throughout their lives, by enabling a growing network of partners to increase the utility of genetic information across the healthcare continuum, and, ultimately, by managing that information on behalf of our customers, enabling improved health and the advancement of molecular medicine around the globe.
As our market share grows, we expect that our business will grow in three stages:
1)Genetic testing: making genetic testing more affordable and more accessible with fast turnaround time. We believe that there is a significant market opportunity for high-volume, low-cost genetic testing that allows us to serve a large number of customers. We launched our first commercial offering in November 2013 with an offering of approximately 200 genes, growing the test menu over time to include more than 20,000 genes to help diagnose disease, inform family planning, and serve healthy individuals. In 2020, we processed billable volume of approximately 659,000 units and generated revenue of $279.6 million reflecting an approximate 41% and 29% increase over 2019 billable volume and revenue, respectively.
4
2)Genome network: sharing genetic information on a global scale to advance science and medicine. We are focusing our efforts on partnering with patients, family members, healthcare professionals, payers, industry professionals, researchers, and clinical trial sponsors to advance the development of our genome network. Our goal is to enable and build a network through which individuals and organizations can access, aggregate, and customize genetic information in order to participate in research, clinical trials, treatment planning, or other related purposes that may benefit the individual and/or their clinician. Individuals can also share information if they feel it will benefit them or will contribute more broadly to furthering knowledge about their conditions.
In addition to investing in informatics solutions and infrastructure to support network development, we have been expanding our partnerships, which now number more than 100 of the world's leading biopharmaceutical companies supporting improved patient diagnosis, clinical trial recruitment and other research-related initiatives. Our biopharmaceutical industry partnerships are complemented by partnerships with leading health systems, executive health programs and leading research institutions, including The Christ Hospital Health Network, the Cleveland Clinic, the Geisinger Health System, the Mayo Clinic, Memorial Sloan Kettering Cancer Center, MedCan, and Stanford Health Care, among others.
Through our recent acquisition of ArcherDX, we partner with global biopharmaceutical companies such as AstraZeneca AB (Publ), Illumina and Merck KGaA, Darmstadt, Germany through collaboration agreements to bring new treatment options for patients to market faster by enabling clinical research and trials.
3)Genome management: building a secure and trusted genome management infrastructure. By generating and storing large amounts of individualized genetic information for every patient sample and enabling the analysis of that cumulative data for broad health research applications, we believe we can create value for all the constituents of our testing platform and partner network. Broad access to centralized, standardized genomic data can benefit patients and their families with information that will improve therapy and outcomes, while it is also expected to aid in the compression of drug development timelines and the greater application of fact-based healthcare decisions throughout life.
Competition
Our competitors include companies that offer molecular genetic testing and consulting services, including specialty and reference laboratories that offer traditional single- and multi-gene tests and biopharmaceutical companies. Principal competitors include companies such as Ambry Genetics, a subsidiary of Konica Minolta Inc.; Athena Diagnostics and Blueprint Genetics, subsidiaries of Quest Diagnostics Incorporated; Baylor-Miraca Genetics Laboratories; Caris Life Sciences, Inc.; Centogene AG; Color Genomics, Inc.; Connective Tissue Gene Test LLC, a subsidiary of Health Network Laboratories, L.P.; Cooper Surgical, Inc.; Emory Genetics Laboratory, a subsidiary of Eurofins Scientific; Foundation Medicine, a subsidiary of Roche Holding AG; Fulgent Genetics, Inc.; GeneDx, a subsidiary of OPKO Health, Inc.; Guardant Health, Inc.; Integrated Genetics, Sequenom Inc., Correlagen Diagnostics, Inc., and MNG Laboratories, subsidiaries of Laboratory Corporation of America Holdings; Myriad Genetics, Inc.; Natera, Inc.; Perkin Elmer, Inc.; PreventionGenetics, LLC; Progenity, Inc.; and Sema4 Genomics; as well as other commercial and academic labs.
In addition, there are a large number of new entrants into the market for genetic information ranging from informatics and analysis pipeline developers to focused, integrated providers of genetic tools and services for health and wellness, including Illumina, Inc. which is also one of our suppliers. In addition to the companies that currently offer traditional genetic testing services and research centers, other established and emerging healthcare, information technology and service companies may commercialize competitive products including informatics, analysis, integrated genetic tools and services for health and wellness.
5
We believe the principal competitive factors in our market are:
•breadth and depth of content;
•quality;
•reliability;
•accessibility of results;
•turnaround time of testing results;
•price and quality of tests;
•coverage and reimbursement arrangements with third-party payers;
•convenience of testing;
•brand recognition of test provider;
•additional value-added services and informatics tools;
•client service; and
•quality of website content.
We believe that we compare favorably with our competitors on the basis of these factors. However, certain competitors and potential competitors have longer operating histories, larger customer bases, greater brand recognition and market penetration, substantially greater financial, technological and research and development resources and selling and marketing capabilities, and more experience dealing with third-party payers. As a result, they may be able to respond more quickly to changes in customer requirements, devote greater resources to the development, promotion and sale of their tests, or sell their tests at prices designed to win significant levels of market share. We may not be able to compete effectively against these organizations.
Regulation
Reimbursement
In April 2014, Congress passed the Protecting Access to Medicare Act of 2014, or PAMA, which included substantial changes to the way in which clinical laboratory services are paid under Medicare. Under PAMA (as amended by the Further Consolidated Appropriations Act, 2020 and the Coronavirus Aid, Relief, and Economic Security (CARES) Act) and its implementing regulations, laboratories that realize at least $12,500 in Medicare Clinical Laboratory Fee Schedule, or CLFS, revenues during the six month reporting period and that receive the majority of their Medicare revenue from payments made under the CLFS or the Physician Fee Schedule must report, beginning in 2017, and then in 2022 and every three years thereafter (or annually for “advanced diagnostic laboratory tests”), private payer payment rates and volumes for their tests. We do not believe that our tests meet the current definition of advanced diagnostic laboratory tests, and therefore believe we are required to report private payer rates for our tests on an every three years basis starting next in 2022. CMS uses the rates and volumes reported by laboratories to develop Medicare payment rates for the tests equal to the volume‑weighted median of the private payer payment rates for the tests. Laboratories that fail to report the required payment information may be subject to substantial civil money penalties.
As set forth under the regulations implementing PAMA, for tests furnished on or after January 1, 2018, Medicare payments for clinical diagnostic laboratory tests are paid based upon these reported private payer rates. For clinical diagnostic laboratory tests that are assigned a new or substantially revised code, initial payment rates for clinical diagnostic laboratory tests that are not advanced diagnostic laboratory tests will be assigned by the cross‑walk or gap‑fill methodology, as under prior law. Initial payment rates for new advanced diagnostic laboratory tests will be based on the actual list charge for the laboratory test.
The payment rates calculated under PAMA went into effect starting January 1, 2018. Where applicable, reductions to payment rates resulting from the new methodology are limited to 10% per test per year in each of the years 2018 through 2020. Rates will be held at 2020 levels during 2021, and then, where applicable based upon median private payer rates reported in 2017 or 2022, reduced by up to 15% per test per year in each of 2022 through 2024 (with a second round of private payer rate reporting in 2022 to establish rates for 2023 through 2025).
PAMA codified Medicare coverage rules for laboratory tests by requiring any local coverage determination to be made following the local coverage determination process. PAMA also authorizes CMS to consolidate coverage policies for clinical laboratory tests among one to four laboratory-specific MACs. These same contractors may also be designated to process claims if CMS determines that such a model is appropriate. It is unclear whether CMS will proceed with contractor consolidation under this authorization.
6
PAMA also authorized the adoption of new, temporary billing codes and/or unique test identifiers for FDA-cleared or approved tests as well as advanced diagnostic laboratory tests. The American Medical Association has created a new section of billing codes, Proprietary Laboratory Analyses (PLA), to facilitate implementation of this section of PAMA. These codes may apply to one or more of our tests if we apply for PLA coding.
In March 2018, CMS published a national coverage determination, or NCD, for next generation sequencing, or NGS tests for somatic (acquired) cancer testing. CMS subsequently updated this NCD in January 2020 to address coverage for NGS tests for germline (inherited) cancer testing and to clarify certain aspects of Medicare’s coverage of NGS for somatic cancer testing. For somatic cancer testing, the updated NCD establishes full coverage for FDA-approved or FDA-cleared NGS-based companion diagnostic assays that report results using report templates that specify treatment options when offered for their FDA-approved or FDA-cleared use(s), ordered by the patient’s treating physician for Medicare beneficiaries with advanced cancer (recurrent, relapsed, refractory, metastatic, or advanced stage III or IV cancer) who have not have previously been tested with the same test using NGS for the same cancer genetic content, and have decided to seek further cancer treatment (e.g., therapeutic chemotherapy.) The NCD also gives MACs the authority to establish local coverage for NGS-based somatic cancer assays that are not FDA-approved or FDA-cleared companion diagnostics when offered to patients meeting the above-referenced criteria. It appears that NGS-based somatic cancer tests provided for patients with cancer that do not meet the above-referenced criteria - e.g., patients with earlier stage cancers - are currently nationally non-covered under the NCD.
Effective January 27, 2020, the NCD also establishes full coverage for FDA-approved or FDA-cleared NGS-based germline tests that report results using report templates that specify treatment options when ordered by the patient’s treating physician for patients with ovarian or breast cancer, a clinical indication for germline testing for hereditary breast or ovarian cancer, and a risk factor for germline breast or ovarian cancer, provided the patient has not previously been tested with the same germline test using NGS for the same germline genetic content. The NCD also gives MACs the authority to establish local coverage for NGS-based germline tests for ovarian or breast cancer that are not FDA-approved or FDA-cleared, as well as for NGS-based tests for any other cancer diagnosis (regardless of the test’s FDA regulatory status) when offered to patients meeting the above-referenced criteria for germline testing. Since we already have local coverage for our germline tests for ovarian and breast cancer, we believe that the NCD will not have a material impact on which of our tests will be reimbursable by CMS for Medicare patients.
Clinical Laboratory Improvement Amendments of 1988, or CLIA
Our clinical reference laboratories in California and Colorado are required to hold certain federal certificates to conduct our business. Under CLIA, we are required to hold certificates applicable to the type of laboratory examinations we perform and to comply with standards covering personnel, facilities administration, inspections, quality control, quality assurance and proficiency testing.
We have current certifications under CLIA to perform testing at our laboratory locations in San Francisco and Irvine, California and Golden, Colorado. To renew our CLIA certifications, we are subject to survey and inspection every two years to assess compliance with program standards. Moreover, CLIA inspectors may make random inspections of our clinical reference laboratories. The regulatory and compliance standards applicable to the testing we perform may change over time, and any such changes could have a material effect on our business.
If our clinical reference laboratories are out of compliance with CLIA requirements, we may be subject to sanctions such as suspension, limitation or revocation of our CLIA certificates, as well as directed plan of correction, state on‑site monitoring, significant civil money penalties, civil injunctive suit or criminal penalties. We must maintain CLIA compliance and certifications to be eligible to bill for diagnostic services provided to Medicare and Medicaid beneficiaries. If we were to be found out of compliance with CLIA requirements and subjected to sanction, our business could be harmed.
7
Laboratory licensure requirements
We are required to maintain in-state licenses to conduct testing in California and Washington. California and Washington laws establish standards for day‑to‑day operations of our laboratories in San Francisco and Irvine, and Seattle, respectively. Such laws mandate proficiency testing, which involves testing of specimens that have been specifically prepared for the laboratories. If our clinical reference laboratories are out of compliance with applicable standards, the appropriate state agency may suspend, restrict or revoke our licenses to operate our clinical reference laboratories, assess substantial civil money penalties, or impose specific corrective action plans. Any such actions could materially affect our business. We maintain current licenses in good standing. However, we cannot provide assurance that state regulators will at all times in the future find us to be in compliance with all such laws.
Several states require the licensure of out‑of‑state laboratories that accept specimens from those states. Our California laboratories hold the required out‑of‑state laboratory licenses for Maryland, New York, Pennsylvania, and Rhode Island. Our Seattle laboratory holds the required out-of-state laboratory licenses in Maryland, New York, Pennsylvania, and Rhode Island (but not California). Our laboratory in Golden, Colorado holds the required out‑of‑state laboratory licenses for California, Maryland, Pennsylvania and Rhode Island (but not New York).
In addition to having laboratory licenses in New York, our clinical reference laboratories are also required to obtain approval on a test‑specific basis for the tests they run as LDTs by the New York State Department of Health, or NYDOH, before specific testing is performed on samples from New York.
Other states may adopt similar licensure requirements in the future, which may require us to modify, delay or stop our operations in such jurisdictions. Complying with licensure requirements in new jurisdictions may be expensive, time‑consuming, and subject us to significant and unanticipated delays. If we identify any other state with such requirements, or if we are contacted by any other state advising us of such requirements, we intend to follow instructions from the state regulators as to how we should comply with such requirements.
We may also be subject to regulation in foreign jurisdictions as we seek to expand international utilization of our tests or such jurisdictions adopt new licensure requirements, which may require review of our tests in order to offer them or may have other limitations such as restrictions on the transport of human blood or saliva necessary for us to perform our tests that may limit our ability to make our tests available outside of the United States.
Federal oversight of laboratory developed tests
We provide many of our tests as laboratory‑developed tests, or LDTs. CMS and certain state agencies regulate the performance of LDTs (as authorized by CLIA and state law, respectively).
Historically, the U.S. Food and Drug Administration, or FDA, has exercised enforcement discretion with respect to most LDTs and has not required laboratories that furnish LDTs to comply with the agency’s requirements for medical devices (e.g., establishment registration, device listing, quality systems regulations, premarket clearance or premarket approval, and post‑ market controls). In recent years, however, the FDA has stated it intends to end its policy of general enforcement discretion and regulate certain LDTs as medical devices. To this end, on October 3, 2014, the FDA issued two draft guidance documents, entitled “Framework for Regulatory Oversight of Laboratory Developed Tests (LDTs)” and “FDA Notification and Medical Device Reporting for Laboratory Developed Tests (LDTs),” respectively, that set forth a proposed risk‑based regulatory framework that would apply varying levels of FDA oversight to LDTs. The FDA has indicated that it does not intend to modify its policy of enforcement discretion until the draft guidance documents are finalized. Subsequently, on January 13, 2017, the FDA published a “discussion paper” in which the agency outlined a substantially revised “possible approach” to the oversight of LDTs. The discussion paper explicitly states that it is not a final version of the 2014 draft guidance and that it does not represent the agency’s “formal position;” rather, the discussion paper describes the evolution of the agency’s thinking on LDTs, which the agency posted to “spur further dialogue.” Notably, in the discussion paper, the agency expressed its willingness to consider “grandfathering” currently marketed LDTs from most or all FDA regulatory requirements.
In August 2020, the U.S. Department of Health and Human Services – the parent agency for FDA – announced that the FDA “will not require premarket review of [LDTs] absent notice-and-comment rulemaking, as opposed to through guidance documents, compliance manuals, website statements, or other informal issuances.” It is unclear at this time whether the Biden Administration will rescind or reverse this policy.
8
It is unclear at this time when, or if, the FDA will finalize its plans to end enforcement discretion (e.g., via notice and comment rulemaking or otherwise), and even then, the new regulatory requirements are expected to be phased‑in over time. Nevertheless, the FDA may attempt to regulate certain LDTs on a case‑by‑case basis at any time.
Legislative proposals addressing the FDA’s oversight of LDTs have been introduced in previous Congresses, and we expect that new legislative proposals will be introduced from time‑to‑time. The likelihood that Congress will pass such legislation and the extent to which such legislation may affect the FDA’s plans to regulate certain LDTs as medical devices is difficult to predict at this time.
If the FDA ultimately regulates certain LDTs, whether via final guidance, final regulation, or as instructed by Congress, our tests may be subject to certain additional regulatory requirements. Complying with the FDA’s requirements can be expensive, time‑consuming, and subject us to significant or unanticipated delays. Insofar as we may be required to obtain premarket clearance or approval to perform or continue performing an LDT, we cannot assure you that we will be able to obtain such authorization. Even if we obtain regulatory clearance or approval where required, such authorization may not be for the intended uses that we believe are commercially attractive or are critical to the commercial success of our tests. As a result, the application of the FDA’s requirements to our tests could materially and adversely affect our business, financial condition, and results of operations.
Notwithstanding the FDA’s current position with respect to oversight of our tests, we may voluntarily decide to pursue FDA pre‑market review for our current tests and/or tests we may offer in the future if we determine that doing so would be appropriate from a strategic perspective – e.g., if CMS indicated that it no longer intended to cover tests offered as LDTs.
Failure to comply with applicable FDA regulatory requirements may trigger a range of enforcement actions by the FDA including warning letters, civil monetary penalties, injunctions, criminal prosecution, recall or seizure, operating restrictions, partial suspension or total shutdown of operations, and denial of or challenges to applications for clearance or approval, as well as significant adverse publicity.
In addition, in November 2013, the FDA issued final guidance regarding the distribution of products labeled for research use only. Certain of the reagents and other products we use in our tests are labeled as research use only products. Certain of our suppliers may cease selling research use only products to us and any failure to obtain an acceptable substitute could significantly and adversely affect our business, financial condition and results of operations.
Medical device regulatory framework
Pursuant to its authority under the Federal Food, Drug, and Cosmetic Act, or FDCA, the FDA has jurisdiction over medical devices, which are defined to include, among other things, in vitro diagnostics, or IVDs. The FDA regulates the research, design, development, pre-clinical and clinical testing, manufacturing, safety, effectiveness, packaging, labeling, storage, recordkeeping, pre-market clearance or approval, adverse event reporting, marketing, promotion, sales, distribution and import and export of medical devices. Specifically, for the test we offer that FDA currently regulates as a device, and if the FDA begins to actively regulate LDTs, then for those tests as well, each new or significantly modified test we seek to commercially distribute in the United States could require either a premarket notification to the FDA requesting permission for commercial distribution under Section 510(k) of the FDCA, also referred to as a 510(k) clearance, or approval from the FDA of a premarket approval, or PMA, application, unless an exemption applies. Both the 510(k) clearance and PMA processes can be resource intensive, expensive, and lengthy, and require payment of significant user fees.
Device classification
Under the FDCA, medical devices are classified into one of three classes (Class I, Class II or Class III) depending on the degree of risk associated with each medical device and the extent of control needed to provide reasonable assurances with respect to safety and effectiveness.
Class I includes devices with the lowest risk to the patient and are those for which safety and effectiveness can be reasonably assured by adherence to General Controls for Medical Devices, which require compliance with the applicable portions of the FDA’s Quality System Regulation, facility registration and product listing, reporting of adverse events and malfunctions, and appropriate, truthful and non-misleading labeling and promotional materials. While some Class I devices also require premarket clearance by the FDA through the 510(k) premarket notification process described below, most Class I products are exempt from the premarket notification requirements.
9
Class II devices are those that are subject to the General Controls, as well as Special Controls as deemed necessary by the FDA to ensure the safety and effectiveness of the device. These Special Controls can include performance standards, patient registries, FDA guidance documents and postmarket surveillance. Most Class II devices are subject to premarket review and clearance by the FDA. Premarket review and clearance by the FDA for Class II devices is accomplished through the 510(k) premarket notification process.
Class III devices include devices deemed by the FDA to pose the greatest risk, such as life-supporting, life-sustaining devices, or implantable devices, in addition to those deemed novel and not substantially equivalent to a legally-marketed predicate device. The safety and effectiveness of Class III devices cannot be reasonably assured solely by the General Controls and Special Controls described above. Therefore, these devices are subject to the PMA process, which is generally more costly and time-consuming than the 510(k) process. Through the PMA process, the applicant must submit data and information demonstrating reasonable assurance of the safety and effectiveness of the device for its intended use to the FDA’s satisfaction. Accordingly, a PMA typically includes, but is not limited to, extensive technical information regarding device design and development, pre-clinical and clinical trial data, manufacturing information, labeling and financial disclosure information for the clinical investigators in device studies. The PMA application must provide valid scientific evidence that demonstrates to the FDA’s satisfaction a reasonable assurance of the safety and effectiveness of the device for its intended use.
The 510(k) clearance process
Under the 510(k) clearance process, the manufacturer must submit to the FDA a premarket notification, demonstrating that the device is “substantially equivalent” to a legally marketed predicate device. A predicate device is a legally marketed device that is not subject to a PMA, i.e., a device that was legally marketed prior to May 28, 1976 (pre-amendments device) and for which a PMA is not required, a device that has been reclassified from Class III to Class II or I, or a device that was previously found substantially equivalent through the 510(k) process. To be “substantially equivalent,” the proposed device must have the same intended use as the predicate device, and either have the same technological characteristics as the predicate device or have different technological characteristics, but the information submitted demonstrates that the device is as safe and effective and does not raise different questions of safety or effectiveness than the predicate device. Clinical data is sometimes required to support substantial equivalence.
After a 510(k) premarket notification is submitted, the FDA determines whether to accept it for substantive review. If it lacks necessary information for substantive review, the FDA will refuse to accept the 510(k) premarket notification. If it is accepted for filing, the FDA begins a substantive review. By statute, the FDA is required to complete its review of a 510(k) notification within 90 days of receiving the 510(k) notification. As a practical matter, clearance often takes longer, and clearance is never assured. Although many 510(k) premarket notifications are cleared without clinical data, the FDA may require further information, including data from samples collected in a clinical setting, to make a determination regarding substantial equivalence, which may significantly prolong the review process. If the FDA agrees that the device is substantially equivalent, it will grant clearance to commercially market the device.
If the FDA determines that the device is not “substantially equivalent” to a predicate device, or if the device is automatically classified into Class III because there is no available predicate device, the device sponsor must then fulfill the much more rigorous premarketing requirements of the PMA approval process, or seek reclassification of the device through the De Novo classification process. The De Novo classification process is an alternate pathway to classify medical devices that are automatically classified into Class III but which are low to moderate risk. A manufacturer can submit a petition for direct de novo review if the manufacturer is unable to identify an appropriate predicate device and the new device or new use of the device presents moderate or low risk. De Novo classification may also be available after receipt of a “not substantially equivalent” letter following submission of a 510(k) to FDA.
10
After a device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a new or major change in its intended use, will require a new 510(k) clearance or, depending on the modification, could require a PMA application. The FDA requires each manufacturer to determine whether the proposed change requires a new submission in the first instance, but the FDA can review any such decision and disagree with a manufacturer’s determination. Many minor modifications are accomplished by an internal letter-to-file in which the manufacturer documents its reasoning for why a change does not require premarket submission to the FDA. The letter-to-file is in lieu of submitting a new 510(k) to obtain clearance for such change. The FDA can always review these letters to file in an inspection. If the FDA disagrees with a manufacturer’s determination regarding whether a new premarket submission is required for the modification of an existing 510(k)-cleared device, the FDA can require the manufacturer to cease marketing and/or recall the modified device until 510(k) clearance or approval of a PMA application is obtained. In addition, in these circumstances, the FDA can impose significant regulatory fines or penalties for failure to submit the requisite application(s).
The PMA approval process
Following receipt of a PMA application, the FDA conducts an administrative review to determine whether the application is sufficiently complete to permit a substantive review. If it is not, the agency will refuse to file the PMA. If it is, the FDA will accept the application for filing and begin the review. The FDA has 180 days to review a filed PMA application, although the review of an application more often occurs over a significantly longer period of time. During this review period, the FDA may request additional information or clarification of information already provided, and the FDA may issue a major deficiency letter to the applicant, requesting the applicant’s response to deficiencies communicated by the FDA.
Before approving or denying a PMA, an FDA advisory committee may review the PMA at a public meeting and provide the FDA with the committee’s recommendation on whether the FDA should approve the submission, approve it with specific conditions, or not approve it. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Prior to approval of a PMA, the FDA may conduct inspections of the clinical trial data and clinical trial sites, as well as inspections of the manufacturing facility and processes. Overall, the FDA review of a PMA application generally takes between one and three years, but may take significantly longer. The FDA can delay, limit or deny approval of a PMA application for many reasons, including:
•the device may not be shown safe or effective to the FDA’s satisfaction;
•the data from pre-clinical studies and/or clinical trials may be found unreliable or insufficient to support approval;
•the manufacturing process or facilities may not meet applicable requirements; and
•changes in FDA clearance or approval policies or adoption of new regulations may require additional data.
If the FDA evaluation of a PMA is favorable, the FDA will issue either an approval letter, or an approvable letter, the latter of which usually contains a number of conditions that must be met in order to secure final approval of the PMA. When and if those conditions have been fulfilled to the satisfaction of the FDA, the agency will issue a PMA approval letter authorizing commercial marketing of the device, subject to the conditions of approval and the limitations established in the approval letter. If the FDA’s evaluation of a PMA application or manufacturing facility is not favorable, the FDA will deny approval of the PMA or issue a not approvable letter. The FDA also may determine that additional tests or clinical trials are necessary, in which case the PMA approval may be delayed for several months or years while the trials are conducted and data is submitted in an amendment to the PMA, or the PMA is withdrawn and resubmitted when the data are available. The PMA process can be expensive, uncertain and lengthy and a number of devices for which the FDA approval has been sought by other companies have never been approved by the FDA for marketing.
New PMA applications or PMA supplements are required for modification to the manufacturing process, equipment or facility, quality control procedures, sterilization, packaging, expiration date, labeling, device specifications, ingredients, materials or design of a device that has been approved through the PMA process. PMA supplements often require submission of the same type of information as an initial PMA application, except that the supplement is limited to information needed to support any changes from the device covered by the approved PMA application and may or may not require as extensive technical or clinical data or the convening of an advisory panel, depending on the nature of the proposed change.
11
In approving a PMA application, as a condition of approval, the FDA may also require some form of post- approval study or post-market surveillance, whereby the applicant conducts a follow-up study or follows certain patient groups for a number of years and makes periodic reports to the FDA on the clinical status of those patients when necessary to protect the public health or to provide additional or longer term safety and effectiveness data for the device. The FDA may also approve a PMA application with other post-approval conditions intended to ensure the safety and effectiveness of the device, such as, among other things, restrictions on labeling, promotion, sale, distribution and use. New PMA applications or PMA supplements may also be required for modifications to any approved diagnostic tests, including modifications to manufacturing processes, device labeling and device design, based on the findings of post-approval studies.
The investigational device process
In the United States, absent certain exceptions, human clinical trials intended to support medical device clearance or approval require an investigational device exemption, or IDE, application. Investigations that meet certain requirements – i.e., involve tests that are labeled investigational use only (IUO), are noninvasive, do not require an invasive sampling procedure that presents significant risk, do not by design or intention introduce energy into a subject, and are not used as a diagnostic procedure without confirmation of the diagnosis by another, medically established product or procedure —are exempt from the IDE requirement. Some types of studies deemed to present “non-significant risk” are deemed to have an approved IDE — without affirmative submission of an IDE application to the FDA — once certain requirements are addressed and Institutional Review Board, or IRB, approval is obtained. If the device presents a “significant risk” to human health, as defined by the FDA, the sponsor must submit an IDE application to the FDA and obtain IDE approval prior to commencing the human clinical trials.
Where applicable, the IDE application must be supported by appropriate data, such as animal and laboratory testing results, showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. Generally, clinical trials for a significant risk device may begin once the IDE application is approved by the FDA and the study protocol and informed consent are approved by appropriate IRBs at the clinical trial sites. Submission of an IDE will not necessarily result in the ability to commence clinical trials, and although the FDA’s approval of an IDE allows clinical testing to go forward for a specified number of subjects, it does not bind the FDA to accept the results of the trial as sufficient to prove the product’s safety and efficacy, even if the trial meets its intended success criteria.
Such clinical trials must be conducted in accordance with the FDA’s IDE regulations that govern investigational device labeling, prohibit promotion and specify an array of recordkeeping, reporting and monitoring responsibilities of study sponsors and study investigators. Clinical trials must further comply with good clinical practice regulations for IRB approval and for informed consent and other human subject protections. Required records and reports are subject to inspection by the FDA for any clinical trials subject to FDA oversight. The results of clinical testing may be unfavorable, or, even if the intended safety and efficacy success criteria are achieved, may not be considered sufficient for the FDA to grant marketing approval or clearance of a product. The commencement or completion of any clinical trial may be delayed or halted, or be inadequate to support approval of a PMA application or clearance of a 510(k) premarket notification, for numerous reasons.
The Breakthrough Devices Program is a voluntary program intended to expedite the review, development, assessment and review of certain medical devices that provide for more effective treatment or diagnosis of life-threatening or irreversibly debilitating human diseases or conditions, provided the device also represents breakthrough technology, is one for which no approved or cleared treatment exists, offers significant advantages over existing approved or cleared alternatives, or is one whose availability is in the best interest of patients. All submissions for devices designated as Breakthrough Devices will receive priority review, meaning that the review of the submission is placed at the top of the appropriate review queue and receives additional review resources, as needed. Although Breakthrough Device designation or access to any other expedited program may expedite the development or approval process, it does not change the standards for approval. Access to an expedited program may also be withdrawn by the FDA if it believes that the designation is no longer supported by data from our clinical development program. Additionally, qualification for any expedited review procedure does not ensure that we will ultimately obtain regulatory clearance or approval for such product.
12
Research use only, or RUO
In the United States, products labeled and sold for research use only, and not for the diagnosis or treatment of disease, are sold to a variety of parties, including biopharmaceutical companies, academic institutions and molecular labs. Because such products are not intended for use in clinical practice in diagnostics, and the products cannot include clinical or diagnostic claims, they are exempt from many regulatory requirements otherwise applicable to medical devices. In particular, while the FDA regulations require that RUO products be labeled, “For Research Use Only. Not for use in diagnostic procedures,” the regulations do not otherwise subject such products to the FDA’s pre- and post-market controls for medical devices.
A significant change in the laws governing RUO products or how they are enforced may require a change to our RUO products business model in order to maintain compliance. For instance, in November 2013 the FDA issued a guidance document entitled “Distribution of In Vitro Diagnostic Products Labeled for Research Use Only or Investigational Use Only”, or the RUO Guidance, which highlights the FDA’s interpretation that distribution of RUO products with any labeling, advertising or promotion that suggests that clinical laboratories can validate the test through their own procedures and subsequently offer it for clinical diagnostic use as a laboratory developed test is in conflict with RUO status. The RUO Guidance further articulates the FDA’s position that any assistance offered in performing clinical validation or verification, or similar specialized technical support, to clinical laboratories, conflicts with RUO status. If we engage in any activities that the FDA deems to be in conflict with the RUO labeling on the products that we sell, we may be subject to immediate, severe and broad FDA enforcement action that would adversely affect our ability to continue operations selling these products. Accordingly, if the FDA finds that we are distributing RUO products in a manner that is inconsistent with its regulations or guidance, we may be forced to stop distribution of our RUO products until we are in compliance, which would reduce our revenues, increase our costs and adversely affect our business, prospects, results of operations and financial condition. In addition, the FDA’s proposed implementation for a new framework for the regulation of LDTs may negatively impact the LDT market and thereby reduce demand for RUO products. If the FDA requires marketing authorization of our RUO products in the future, there can be no assurance that the FDA will ultimately grant any clearance or approval we request in a timely manner, or at all.
Post-market regulation
After a device is cleared or approved for marketing, numerous and pervasive regulatory requirements continue to apply. These include:
•establishment registration and device listing with the FDA;
•QSR requirements, which require manufacturers, including third-party manufacturers, to follow stringent design, testing, control, documentation and other quality assurance procedures during all aspects of the design and manufacturing process;
•labeling regulations and FDA prohibitions against the promotion of investigational products, or the promotion of “off-label” uses of cleared or approved products;
•requirements related to promotional activities;
•clearance or approval of product modifications to 510(k)-cleared devices that could significantly affect safety or effectiveness or that would constitute a major change in intended use of one of our cleared devices, or approval of certain modifications to PMA-approved devices;
•medical device reporting regulations, which require that a manufacturer report to the FDA if a device it markets may have caused or contributed to a death or serious injury, or has malfunctioned and the device or a similar device that it markets would be likely to cause or contribute to a death or serious injury, if the malfunction were to recur;
•correction, removal and recall reporting regulations, which require that manufacturers report to the FDA field corrections and product recalls or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FDCA that may present a risk to health;
•the FDA’s recall authority, whereby the agency can order device manufacturers to recall from the market a product that is in violation of governing laws and regulations; and
•post-market surveillance activities and regulations, which apply when deemed by the FDA to be necessary to protect the public health or to provide additional safety and effectiveness data for the device.
13
Device manufacturing processes are required to comply with the applicable portions of the QSR, which cover the methods and the facilities and controls for the design, manufacture, testing, production, processes, controls, quality assurance, labeling, packaging, distribution, installation and servicing of finished devices intended for human use. The QSR also requires, among other things, maintenance of a device master file, device history file, and complaint files. Manufacturers are subject to periodic scheduled or unscheduled inspections by the FDA. A failure to maintain compliance with the QSR requirements could result in the shut-down of, or restrictions on, manufacturing operations and the recall or seizure of products. The discovery of previously unknown problems with products, including unanticipated adverse events or adverse events of increasing severity or frequency, whether resulting from the use of the device within the scope of its clearance or approval or off-label by a physician in the practice of medicine, could result in restrictions on the device, including the removal of the product from the market or voluntary or mandatory device recalls.
The FDA has broad regulatory compliance and enforcement powers. If the FDA determines that a manufacturer has failed to comply with applicable regulatory requirements, it can take a variety of compliance or enforcement actions, including the following:
•issuance of warning letters, untitled letters, fines, injunctions, consent decrees and civil penalties;
•requesting or requiring recalls, withdrawals, or administrative detention or seizure of our products;
•imposing operating restrictions or partial suspension or total shutdown of production;
•refusing or delaying requests for 510(k) marketing clearance or PMA approvals of new products or modified products;
•withdrawing 510(k) clearances or PMA approvals that have already been granted;
•refusal to grant export approvals for our products; or
•criminal prosecution.
HIPAA and state privacy, security and breach notification laws
Under the administrative simplification provisions of the Health Insurance Portability and Accountability Act of 1996, or HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH, the U.S. Department of Health and Human Services issued regulations that establish uniform standards governing the conduct of certain electronic healthcare transactions and requirements for protecting the privacy and security of protected health information (PHI) used or disclosed by covered entities, including most health care providers and their respective business associates, as well as the business associates’ subcontractors. Four principal regulations with which we are required to comply have been issued in final form under HIPAA and HITECH: privacy regulations, security regulations, breach notification regulations, and standards for electronic transactions, which establish standards for common healthcare transactions.
The privacy regulations cover the use and disclosure of PHI by covered entities as well as business associates, which are persons or entities that perform certain functions for or on behalf of a covered entity that involve the creation, receipt, maintenance, or transmittal of PHI. Business associates are defined to include a subcontractor to whom a business associate delegates a function, activity, or service, other than in the capacity of the business associate’s workforce. As a general rule, a covered entity or business associate may not use or disclose PHI except as permitted or required under the privacy regulations. The privacy regulations also set forth certain rights that an individual has with respect to his or her PHI maintained by a covered entity or business associate, including the right to access or amend certain records containing his or her PHI, request restrictions on the use or disclosure of his or her PHI, or request an accounting of disclosures of his or her PHI.
Covered entities and business associates also must comply with the security regulations, which establish requirements for safeguarding the confidentiality, integrity, and availability of PHI that is electronically transmitted or electronically stored. In addition, HITECH established, among other things, certain breach notification requirements with which covered entities and business associates must comply. In particular, a covered entity must notify any individual whose unsecured PHI is breached according to the specifications set forth in the breach notification rule. A covered entity must also notify the Secretary of the U.S. Department of Health and Human Services and, under certain circumstances, the media of a breach of unsecured PHI.
14
There are significant civil and criminal penalties that may be imposed on a covered entity or business associate for violating HIPAA. A covered entity or business associate may also be liable for civil money penalties for a violation that is based on an act or omission of any of its agents, including a downstream business associate, as determined according to the federal common law of agency. Additionally, to the extent that we submit electronic healthcare claims and payment transactions that do not comply with the electronic data transmission standards established under HIPAA and HITECH, payments to us may be delayed or denied.
The HIPAA privacy, security, and breach notification regulations establish a uniform federal “floor” and do not supersede state laws that are more stringent or provide individuals with greater rights with respect to the privacy or security of, and access to, their records containing PHI or insofar as such state laws apply to personal information that is broader in scope than PHI as defined under HIPAA. Massachusetts, for example, has a state law that protects the privacy and security of personal information of Massachusetts residents. In addition, every U.S. state has a data breach notification law that requires entities to report certain security breaches to affected consumers and, in some instances, state regulators and consumer reporting agencies. Many states also have laws or regulations that specifically apply to genetic testing and genetic information and are more stringent than the standards under HIPAA. These state genetic information privacy laws include specific informed consent requirements for the conduct of genetic testing and restrict the collection, use, disclosure, or retention of genetic information. Failure to comply with applicable state laws that impose privacy, security, or breach notification requirements for genetic or other personal information could result in significant civil or criminal penalties, administrative actions, or private causes of action by patients, and adversely affect our business, results of operations and reputation.
Federal and state consumer protection laws
The Federal Trade Commission, or FTC, is an independent U.S. law enforcement agency charged with protecting consumers and enhancing competition across broad sectors of the economy. The FTC’s primary legal authority with respect to data privacy and security comes from Section 5 of the FTC Act, which prohibits unfair or deceptive acts or practices in the marketplace. The FTC has increasingly used this broad authority to police data privacy and security, using its powers to investigate and bring lawsuits. Where appropriate, the FTC can seek a variety of remedies, such as but not limited to requiring the implementation of comprehensive privacy and security programs, biennial assessments by independent experts, monetary redress to consumers, and provision of robust notice and choice mechanisms to consumers. In addition to its enforcement mechanisms, the FTC uses a variety of tools to protect consumers’ privacy and personal information, including pursuing enforcement actions to stop violations of law, conducting studies and issuing reports, hosting public workshops, developing educational materials, and testifying before the U.S. Congress on issues that affect consumer privacy.
The vast majority of data privacy cases brought by the FTC fall under the “deceptive” acts prong of Section 5. These cases often involve a failure on the part of a company to adhere to its own privacy and data protection principles set forth in its policies. To avoid Section 5 violations, the FTC encourages companies to build privacy protections and safeguards into relevant portions of their business, and to consider privacy and data protection as the company grows and evolves. In addition, privacy notices should clearly and accurately disclose the type(s) of personal information the company collects, how the company uses and shares that information, and the security measures used by the company to protect that information.
In recent years, the FTC’s enforcement under Section 5 related to data security has included alleged violations of the “unfairness” prong. Many of these cases have alleged that companies were unfair to consumers because they failed to take reasonable and necessary measures to protect consumer data. The FTC has not provided bright line rules defining what constitutes “reasonable and necessary measures” for implementing a cybersecurity program, but it has provided guidance, tips and advice for companies. The FTC has also published past complaints and consent orders, which it urges companies use as guidance to help avoid an FTC enforcement action, even if a data breach or loss occurs.
In addition to the FTC Act, most U.S. states have unfair and deceptive acts and practices statutes, known as UDAP statutes, that substantially mirror the FTC Act and have been applied in the privacy and data security context. These vary in substance and strength from state to state. Many have broad prohibitions against unfair and deceptive acts and practices. These statutes generally allow for private rights of action and are enforced by the states’ Attorneys General.
15
California Consumer Privacy Act
The California Consumer Privacy Act, or CCPA, is a comprehensive consumer privacy law that took effect on January 1, 2020, and regulates how certain for-profit businesses that meet one or more CCPA applicability thresholds collect, use, and disclose the personal information of consumers who reside in California. Among other things, the CCPA confers to California consumers the right to receive notice of the categories of personal information that will be collected by a business, how the business will use and share the personal information, and the third parties who will receive the personal information; the CCPA also confers rights to access, delete, or transfer personal information; and the right to receive equal service and pricing from a business after exercising a consumer right granted by the CCPA. In addition, the CCPA allows California consumers the right to opt out of the “sale” of their personal information, which the CCPA defines broadly as any disclosure of personal information to a third party in exchange for monetary or other valuable consideration. The CCPA also requires a business to implement reasonable security procedures to safeguard personal information against unauthorized access, use, or disclosure.
The CCPA does not apply to personal information that is PHI under HIPAA. The CCPA also does not apply to a HIPAA-regulated entity to the extent that the entity maintains patient information in the same manner as PHI. In addition, California amended the law in September 2020 to clarify that de-identified data as defined under HIPAA will also be exempt from the CCPA. Accordingly, we do not have CCPA compliance obligations with respect to most genetic testing and patient information we collect and process. However, we are required to comply with the CCPA insofar as we collect other categories of California consumers’ personal information, such as information about California employees, contractors, and business-to-business contacts. The CCPA provides partial exemptions for employee and business-to-business information that are set to expire on January 1, 2023. In addition, to the extent that we sell or license de-identified information that is derived from California patients’ information, the contracts for the sale or license of such de-identified information will need to include certain provisions required under the CCPA beginning January 1, 2021.
The California Attorney General has had authority to enforce the CCPA and its implementing regulations against covered businesses since July 1, 2020. The CCPA provides for civil penalties for violations, as well as private right of action for data breaches that result from a business’ failure to implement and maintain reasonable data security procedures.
On November 3, 2020, California passed the California Privacy Rights Act (CPRA) through a ballot initiative. The CPRA will create a new California Privacy Protection Agency, an “independent watchdog” whose mission is both to “vigorously enforce” the CPRA and “ensure that businesses and consumers are well‐informed about their rights and obligations.” Among other things, the CPRA will create a new category of “sensitive personal information” and offer consumers the right to limit processing of such information, impose purpose limitation, data minimization, data retention, and security compliance obligations on regulated businesses, and add or modify the rights available to consumers, including by providing a right to correct the information a business holds about them. The CPRA’s amendments to the CCPA will take effect on January 1, 2023, and will generally apply to personal information collected by businesses on or after January 1, 2022. The California Attorney General will have authority to begin enforcing the CPRA’s amendments to the CCPA beginning on July 1, 2022.
Privacy and data protection laws
There are a growing number of jurisdictions around the globe that have privacy and data protection laws that may apply to Invitae as it enters or expands its business in jurisdictions outside of the United States. These laws are typically triggered by a company’s establishment or physical location in the jurisdiction, data processing activities that take place in the jurisdiction, and/or the processing of personal information about individuals located in that jurisdiction. Certain international privacy and data protection laws, such as those in the European Union (EU), are more restrictive and prescriptive than those in the U.S., while other jurisdictions may have laws less restrictive or prescriptive than those in the U.S. Enforcement of these laws varies from jurisdiction to jurisdiction, with a variety of consequences, including civil or criminal penalties, litigation, private rights of action or damage to our reputation.
16
Europe
The EU’s General Data Protection Regulation, or GDPR, took effect on May 25, 2018. The GDPR applies to any entity established in the EU as well as extraterritorially to any entity outside the EU that offers goods or services to, or monitors the behavior of, individuals who are located in the EU. The GDPR imposes strict requirements on controllers and processors of personal data, including enhanced protections for “special categories” of personal data, which includes sensitive information such as health and genetic information of data subjects. The GDPR also grants individuals various rights in relation to their personal data, including the rights of access, rectification, objection to certain processing and deletion. The GDPR provides an individual with an express right to seek legal remedies if the individual believes his or her rights have been violated. Failure to comply with the requirements of the GDPR or the related national data protection laws of the member states of the EU, which may deviate from or be more restrictive than the GDPR, may result in significant administrative fines issued by EU regulators. Maximum penalties for violations of the EU GDPR are capped at 20 million Euros or 4% of an organization's annual global revenue, whichever is greater.
Australia
Australia’s federal Privacy Act 1988, or the Privacy Act, and the 13 Australian Privacy Principles, or the APPs, contained in the Privacy Act, apply to government agencies and private sector organizations with annual turnover exceeding AU $3 million. The Privacy Act extends to all of Australia's external territories, but also applies to an act done, or practice engaged in, or outside Australia (and Australia's external territories) by an organization, or small business operator, that has a link to Australia, such as a continued presence, partnership, incorporation, central management and control, or citizenship in Australia. An organization may also have a link to Australia if the organization conducts business in Australia and collects or stores personal information in Australia. The Privacy Act applies to any collection, holding, use or disclosure of personal information by a regulated entity, with enhanced protections for sensitive information such as genetic information. The Privacy Act prescribes certain rights for individuals, including rights to know why the information is collected, how it is used, and to whom it is disclosed, the right of the individual not to identify themself in certain circumstances, the right of access, the right to stop receiving unwanted direct marketing, the right to correct information, and the right to make a complaint. Australia’s Privacy Commissioner enforces the Privacy Act and any acts that may violate an individual’s privacy. The Privacy Commissioner can levy significant fines on individuals and corporations that violate the Privacy Act.
Canada
Canada has several federal, provincial and territorial privacy statutes that govern the protection of personal information. The Personal Information Protection and Electronic Documents Act 2000, or PIPEDA, applies to the collection, use, and disclosure of personal information in the course of commercial activities in Canada. Although PIPEDA is silent with respect to its extraterritorial application, the Federal Court of Canada has concluded that PIPEDA applies to businesses established in other jurisdictions if there is a “real and substantial connection” between the organization’s activities and Canada. PIPEDA and provincial data protection laws require specific notices regarding openness and transparency and require regulated organizations to obtain consent in order to process such information. Canadian individuals enjoy rights or access and to correct inaccuracies. Violations of Canadian data protection laws can result in significant fines.
India
The Indian Constitution was recently interpreted to include a fundamental right to privacy. In addition, India’s laws and regulations address specific sectoral data protection concerns. The Information Technology Act 2000, as amended, or the IT Act, is the primary national law regulating the collection and use of personal information that is sensitive. The IT Act applies to corporations and other “body corporates” that possess, maintain, or otherwise process personal information, including body corporates that act on behalf of other body corporates. Certain provisions of the IT Act provide liability for negligent handling of personal information. For example, the IT Act provides that any corporation or other body corporate that handles sensitive personal data is liable to pay damages for any loss caused by its negligence in implementing and maintaining reasonable security practices and procedures.
17
In addition, the Information Technology (Reasonable Security Practices and Procedures and Sensitive Personal Data or Information) Rules 2011, or the Data Privacy Rules, issued under the IT Act regulate the use of personal information and sensitive personal data. The Data Privacy Rules mandate that businesses have a privacy policy, obtain consent when collecting or transferring personal information, and inform the data subject about any recipients of that data. The IT Act includes a private right of action for individuals, and authorizes criminal punishment (with a fine, three years in prison, or both) for disclosing personal information without the consent of the data subject or in breach of any relevant contract.
Israel
Israel’s data protection regime is governed primarily by the Protection of Privacy Law and the regulations promulgated under it, or the PPL, and the guidelines of the Israeli regulator, the Privacy Protection Authority, or the PPA. The PPL applies to: (1) database owners, database holders, and database managers based in Israel; and (2) data processing operations that take place in Israel, regardless of whether the individuals about whom the data relates are residents or citizens of Israel. The PPL could also be interpreted to apply to non-Israeli database owners, database holders, or database managers that process personal information about Israeli residents or citizens when such processing takes place outside of Israel. Various regulations promulgated under the PPL by the PPA set out rules and procedures for data security, data retention, data subject rights, and cross border transfers of data. These regulations also do not clearly state their jurisdictional scope, such that there is a risk they could be interpreted as applying to foreign-based entities that process data about Israeli citizens.
The PPA is required to maintain a registry of databases and is empowered to supervise compliance with and investigate alleged violations of the PPL and related regulations. The PPA may impose administrative fines for violations of the PPL and related regulations, and willful violations may result in criminal liability and up to five years in prison. A breach of privacy is also actionable, and an individual claimant may obtain monetary compensation or injunctive relief. A court may award statutory damages without proof of damages for breach of privacy rights. If the breach was intentional, the damages may be doubled. The PPL also specifies that an act or omission in breach of certain of its provisions, such as failure to ensure data security, may give rise to a tort claim.
Japan
Japan’s primary data protection law, the Act on the Protection of Personal Information, or APPI, was recently amended to include GDPR-like requirements, including additional transparency requirements, data transfer obligations, enhanced data breach notification requirements, additional data subject rights and stronger penalties for violations, including significant fines. The amendment clarifies that its provisions, obligations and penalties apply to entities outside of Japan that supply goods or services in Japan and handle personal information from an individual in Japan.
Information Blocking Prohibition
On May 1, 2020, the Office of the National Coordinator for Health Information Technology promulgated final regulations under the authority of the 21st Century Cures Act to impose new conditions to obtain and maintain certification of certified health information technology and prohibit certain covered actors, including developers of certified health information technology, health information networks / health information exchanges, and health care providers, from engaging in activities that are likely to interfere with the access, exchange, or use of electronic health information (information blocking). The final regulations further defined exceptions for activities that are permissible, even though they may have the effect of interfering with the access, exchange, or use of electronic health information. The information blocking effective date is April 5, 2021. Under the 21st Century Cures Act, health care providers that violate the information blocking prohibition will be subject to appropriate disincentives, which the U.S. Department of Health and Human services has yet to establish through required rulemaking. Developers of certified information technology and health information networks / health information exchanges, however, may be subject to civil monetary penalties of up to $1 million per violation. The U.S. Department of Health and Human Services Office of Inspector General has the authority to impose such penalties and on April 24, 2020 published a proposed rule to codify new authority in regulation, which the agency proposed would be effective 60 days after it issues a final rule, but in no event before November 2, 2020. The U.S. Department of Health and Human Services Office of Inspector General has not yet issued a final rule.
18
Federal, state and foreign fraud and abuse laws
In the United States, there are various fraud and abuse laws with which we must comply, and we are potentially subject to regulation by various federal, state and local authorities, including CMS, other divisions of the U.S. Department of Health and Human Services (e.g., the Office of Inspector General), the U.S. Department of Justice, and individual U.S. Attorney offices within the Department of Justice, and state and local governments. We also may be subject to foreign fraud and abuse laws.
In the United States, the federal Anti-Kickback Statute prohibits knowingly and willfully offering, paying, soliciting or receiving remuneration, directly or indirectly, overtly or covertly, in cash or in kind, to induce or in return for the referral of an individual for the furnishing of or arranging for the furnishing of any item or service for which payment may be made in whole or in part by a federal healthcare program, or the purchasing, leasing, ordering or arranging for or recommending purchasing, leasing or ordering of any good, facility, service or item for which payment may be made in whole or in part by a federal healthcare program. Many courts have held that the Anti-Kickback Statute may be violated if any one purpose of the remuneration is to induce or reward patient referrals or other federal healthcare program business, regardless of whether there are other legitimate purposes for the arrangement. The definition of “remuneration” has been broadly interpreted to include anything of value, including gifts, discounts, credit arrangements, payments of cash, consulting fees, waivers of co-payments, ownership interests, and providing anything at less than its fair market value. The Anti-Kickback Statute is broad and may technically prohibit many innocuous or beneficial arrangements within the healthcare industry. The Anti-Kickback Statute includes several statutory exceptions, and the U.S. Department of Health and Human Services has issued a series of regulatory “safe harbors.” These exceptions and safe harbor regulations set forth certain requirements for various types of arrangements, which, if met, will protect the arrangement from potential liability under the Anti-Kickback Statute. Although full compliance with the statutory exceptions or regulatory safe harbors ensures against liability under the federal Anti-Kickback Statute, the failure of a transaction or arrangement to fit within a specific statutory exception or regulatory safe harbor does not necessarily mean that the transaction or arrangement is illegal or that prosecution under the federal Anti-Kickback Statute will be pursued. Penalties for violations of the Anti-Kickback Statute are severe, and include imprisonment, criminal fines, civil money penalties, and exclusion from participation in federal healthcare programs. Many states also have anti- kickback statutes, some of which may apply to items or services reimbursed by any third-party payer, including commercial insurers.
There are also federal laws related to healthcare fraud and false statements, among others, that apply to healthcare matters. The healthcare fraud statute prohibits, among other things, knowingly and willfully executing a scheme to defraud any healthcare benefit program, including private payers. A violation of this statute is a felony and may result in fines, imprisonment, or exclusion from governmental payer programs such as the Medicare and Medicaid programs. The false statements statute prohibits, among other things, knowingly and willfully falsifying, concealing or covering up a material fact, or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items, or services. A violation of this statute is a felony and may result in fines, imprisonment, or exclusion from governmental payer programs.
Another development affecting the healthcare industry is the increased enforcement of the federal False Claims Act and, in particular, actions brought pursuant to the False Claims Act’s “whistleblower” or “qui tam” provisions. The False Claims Act imposes liability on any person or entity that, among other things, knowingly presents, or causes to be presented, a false or fraudulent claim for payment by a federal governmental payer program. The qui tam provisions of the False Claims Act allow a private individual to bring actions on behalf of the federal government alleging that the defendant has defrauded the federal government by presenting or causing to be presented a false claim to the federal government and permit such individuals to share in any amounts paid by the entity to the government in fines or settlement. When an entity is determined to have violated the False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties for each false claim. For penalties assessed after June 19, 2020, whose associated violations occurred after November 2, 2015, the penalties range from $11,665 to $23,331 for each false claim. The minimum and maximum per claim penalty amounts are subject to annual increases for inflation.
In addition, various states have enacted false claim laws analogous to the federal False Claims Act, and some of these state laws apply where a claim is submitted to any third-party payer and not only a governmental payer program.
19
Additionally, the civil monetary penalties statute imposes penalties against any person or entity that, among other things, is determined to have knowingly presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or for a claim that is false or fraudulent. This law also prohibits the offering or transfer of remuneration to a Medicare or state healthcare program beneficiary if the person knows or should know it is likely to influence the beneficiary’s selection of a particular provider, practitioner, or supplier for items or services reimbursable by Medicare or a state healthcare program. There are several exceptions to the prohibition on beneficiary inducement.
The Eliminating Kickbacks in Recovery Act of 2018, or EKRA, prohibits payments for referrals to recovery homes, clinical treatment facilities, and laboratories. EKRA’s reach extends beyond federal health care programs, to include private insurance (i.e., it is an “all payer” statute). For purposes of EKRA, the term “laboratory” is defined broadly and without reference to any connection to substance use disorder treatment. EKRA is a criminal statute and violations can result in fines of up to $200,000, up to 10 years in prison, or both, per violation. The law includes a limited number of exceptions, some of which closely align with corresponding federal Anti-Kickback Statute exceptions and safe harbors and others that materially differ.
We are also subject to the U.S. Foreign Corrupt Practices Act, or FCPA, which prohibits companies and their intermediaries from making payments in violation of law to non-U.S. government officials for the purpose of obtaining or retaining business or securing any other improper advantage. In Europe various countries have adopted anti-bribery laws providing for severe consequences, in the form of criminal penalties and/or significant fines, for individuals and/or companies committing a bribery offence. Violations of these anti-bribery laws, or allegations of such violations, could have a negative impact on our business, results of operations and reputation. For instance, in the United Kingdom, under the Bribery Act 2010, which went into effect in July 2011, a bribery occurs when a person offers, gives or promises to give a financial or other advantage to induce or reward another individual to improperly perform certain functions or activities, including any function of a public nature. Bribery of foreign public officials also falls within the scope of the Bribery Act 2010. Under the new regime, an individual found in violation of the Bribery Act 2010, faces imprisonment of up to ten years. In addition, the individual can be subject to an unlimited fine, as can commercial organizations for failure to prevent bribery.
The Physician Payments Sunshine Act, enacted as part of the Affordable Care Act, also imposed annual reporting requirements on entities including manufacturers of certain devices, medical supplies, drugs and biologics for certain payments and transfers of value that the manufacturer provides, directly or indirectly, to or on behalf of certain types of health care providers, including physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, as defined by such law as well as ownership and investment interests held by physicians and their immediate family members. Beginning in 2022, applicable manufacturers also will be required to report such information regarding its relationships with physician assistants, nurse practitioners, clinical nurse specialists, certified registered nurse anesthetists and certified nurse midwives during the previous year. The Physician Payments Sunshine Act also requires entities including applicable manufacturers to report certain ownership and investment interests held by such physicians and their immediate family members in such manufacturers. In addition, certain states, such as Vermont and Massachusetts, have enacted laws that impose certain reporting requirements for payments and transfers of value provided to covered healthcare providers. These state laws are not preempted by the federal Physician Payments Sunshine Act to the extent the state law requires the reporting of information that is not required to be reported under the federal Physician Payments Sunshine Act. Finally, certain states such as Massachusetts, Nevada, and Vermont have enacted laws that limit or prohibit the provision of payments or other transfers of value to covered recipients, such as certain health care providers, hospitals, and health benefit plan administrators.
20
Physician referral prohibitions
A federal law directed at “self-referrals,” commonly known as the “Stark Law,” prohibits a physician from referring a patient to an entity for certain Medicare-covered designated health services, including laboratory services, if the physician, or an immediate family member, has a financial relationship with the entity, unless an exception applies. The Stark Law also prohibits an entity from billing for services furnished pursuant to a prohibited referral. A physician or entity that engages in a scheme to circumvent the Stark Law’s referral prohibition may be fined up to $172,137 for each such arrangement or scheme. In addition, any person who presents or causes to be presented a claim to the Medicare program in violation of the Stark Law is subject to civil monetary penalties of up to $25,820 per service, an assessment of up to three times the amount claimed and possible exclusion from participation in federal healthcare programs. Bills submitted in violation of the Stark Law may not be paid by Medicare, and any person collecting any amounts with respect to any such prohibited bill is obligated to refund such amounts. Many states have comparable laws that apply to services covered by other third-party payers. The Stark Law also prohibits state receipt of federal Medicaid matching funds for services furnished pursuant to a prohibited referral. This provision of the Stark Law has not been implemented by regulations, but some courts have held that the submission of claims to Medicaid that would be prohibited as self-referrals under the Stark Law for Medicare could implicate the False Claims Act.
Corporate practice of medicine
Numerous states have enacted laws prohibiting business corporations, such as us, from practicing medicine and employing or engaging clinicians to practice medicine, generally referred to as the prohibition against the corporate practice of medicine. These laws are designed to prevent interference in the medical decision-making process by anyone who is not a licensed physician. For example, California’s Medical Board has indicated that determining what diagnostic tests are appropriate for a particular condition and taking responsibility for the ultimate overall care of the patient, including providing treatment options available to the patient, would constitute the unlicensed practice of medicine if performed by an unlicensed person. Violation of these corporate practice of medicine laws may result in civil or criminal fines, as well as sanctions imposed against us and/or the professional through licensure proceedings.
Intellectual property
We rely on a combination of intellectual property rights, including trade secrets, copyrights, trademarks, customary contractual protections and, to a lesser extent, patents, to protect our core technology and intellectual property. With respect to patents, we believe that the practice of patenting individual genes, along with patenting tools and methods specific to individual genes, has impeded the progress of the genetic testing industry beyond single gene tests and is antithetical to our core principle that patients should own and control their own genomic information. The U.S. Supreme Court has issued a series of unanimous (9-0) decisions setting forth limits on the patentability of natural phenomena, natural laws, abstract ideas and their applications — i.e., Mayo Collaborative v. Prometheus Laboratories (2012), or Mayo, Association for Molecular Pathology v. Myriad Genetics (2013), or Myriad, and Alice Corporation v. CLS Bank (2014), or Alice. As discussed below, we believe the Mayo, Myriad and Alice decisions bring clarity to the limits to which patents may cover specific genes, mutations of such genes, or gene-specific technology for determining a patient’s genomic information.
Patents
U.S. Supreme Court cases have clarified that naturally occurring DNA sequences are natural phenomena, which should not be patentable. On June 13, 2013, the U.S. Supreme Court decided Myriad, a case challenging the validity of patent claims held by Myriad relating to the cancer genes BRCA1 and BRCA2. The Myriad Court held that genomic DNAs that have been isolated from, or have the same sequence as, naturally occurring samples, such as the DNA constituting the BRCA1 and BRCA2 genes or fragments thereof, are not eligible for patent protection. Instead, the Myriad Court held that only those complementary DNAs (cDNAs) which have a sequence that differs from a naturally occurring fragment of genomic DNA may be patent eligible. Because it will be applied by other courts to all gene patents, the holding in Myriad also invalidates patent claims to other genes and gene variants. Prior to Myriad, on August 16, 2012, the U.S. Court of Appeals for the Federal Circuit had held that certain patent claims of Myriad directed to methods of comparing or analyzing BRCA1 and BRCA2 sequences to determine whether or not a person has a variant or mutation are unpatentable abstract processes, and Myriad did not appeal such ruling.
21
We do not currently have any patents or patent applications directed to the sequences of specific genes or variants of such genes, nor do we rely on any such in-licensed patent rights of any third party. We believe that correlations between specific gene variants and a person’s susceptibility to certain conditions or diseases are natural laws that are not patentable under the U.S. Supreme Court’s decision in Mayo. The Mayo case involved patent claims directed to optimizing, on a patient-specific basis, the dosage of a certain drug by measuring its metabolites in a patient. The Mayo Court determined that patent claims directed at detection of natural correlations, such as the correlation between drug metabolite levels in a patient and that drug’s optimal dosage for such patient, are not eligible for patent protection. The Mayo Court held that claims based on this type of comparison between an observed fact and an understanding of that fact’s implications represent attempts to patent a natural law and, moreover, when the processes for making the comparison are not themselves sufficiently inventive, claims to such processes are similarly patent-ineligible. On June 19, 2014, the U.S. Supreme Court decided Alice, where it amplified its Mayo and Myriad decisions and clarified the analytical framework for distinguishing between patents that claim laws of nature, natural phenomena and abstract ideas and those that claim patent-eligible applications of such concepts. According to the Alice Court, the analysis depends on whether a patent claim directed to a law of nature, a natural phenomenon or an abstract idea contains additional elements, an “inventive concept,” that “is sufficient to ensure that the patent in practice amounts to significantly more than a patent upon the [ineligible concept] itself;” (citing Mayo).
We believe that Mayo, Myriad and Alice not only render as unpatentable genes, gene fragments and the detection of a person’s sequence for a gene, but also have the same effect on generic applications of conventional technology to specific gene sequences. For example, we believe that generic claims to primers or probes directed to specific gene sequences and uses of such primers and probes in determining a person’s genetic information are not patentable. We do not currently have any patents or patent applications directed to such subject matter nor have we in-licensed such patents rights of any third party.
Unlike patents directed to specific genes, we do rely upon, in part, patent protection to protect technology that is not gene-specific and that provides us with a potential competitive advantage as we focus on making comprehensive genetic information less expensive and more broadly available to our customers. In this regard, we have issued U.S. patents, pending U.S. patent applications and corresponding non-U.S. patents and patent applications directed to various aspects of our laboratory, analytic and business practices. We intend to pursue further patent protection where appropriate.
For information regarding legal actions that pertain to intellectual property rights, see Note 8, “Commitments and contingencies” in Notes to Consolidated Financial Statements in Part II, Item 8 of this report.
Trade secrets
In addition to seeking patent protection for some of our laboratory, analytic and business practices, we also rely on trade secrets, including unpatented know-how, technology and other proprietary information, to maintain and develop our competitive position. We have developed proprietary procedures for both the laboratory processing of patient samples and the analysis of the resulting data to generate clinical reports. For example, we have automated aspects of our processes for curating information about known variants, identifying variants in an individual’s sequence information, associating those variants with known information about their potential effects on disease, and presenting that information for review by personnel responsible for its interpretation and for the delivery of test reports to clinicians and patients. We try to protect these trade secrets, in part, by taking reasonable steps to keep them confidential. This includes entering into nondisclosure and confidentiality agreements with parties who have access to them, such as our employees and certain third parties. We also enter into invention or patent assignment agreements with our employees and consultants that obligate them to assign to us any inventions developed in the course of their work for us. However, we may not enter into such agreements with all relevant parties, and these parties may not abide by the terms of their agreements. Despite measures taken to protect our intellectual property, unauthorized parties might copy or independently develop and commercially exploit aspects of our technology or obtain and use information that we regard as proprietary.
Trademarks
We work hard to achieve a high level of quality in our operations and to provide our customers with a superior experience when interacting with us. As a consequence, our brand is very important to us, as it is a symbol of our reputation and representative of the goodwill we seek to generate with our customers. As a consequence, we have invested significant resources in protection of our trademarks.
22
Environmental matters
Our operations require the use of hazardous materials (including biological materials) that subject us to a variety of federal, state and local environmental and safety laws and regulations. Some of these regulations provide for strict liability, holding a party potentially liable without regard to fault or negligence. We could be held liable for damages and fines as a result of our, or others’, business operations should contamination of the environment or individual exposure to hazardous substances occur. We cannot predict how changes in laws or new regulations will affect our business, operations or the cost of compliance.
Raw materials and suppliers
We rely on a limited number of suppliers, or, in some cases, sole suppliers, including Agena Bioscience, Inc., Illumina, Inc., Integrated DNA Technologies Incorporated, Roche Holdings Ltd., QIAGEN, Inc. and Twist Bioscience Corporation for certain laboratory reagents, as well as sequencers and other equipment and materials which we use in our laboratory operations. We are in active litigation with affiliates of QIAGEN, Inc. as described in Note 8, "Commitments and contingencies" in Notes to Consolidated Financial Statements in Part II, Item 8 of this report. We rely on Illumina as the sole supplier of next generation sequencers and associated reagents and as the sole provider of maintenance and repair services for these sequencers and QIAGEN, Inc. to provide the enzymes that we use in our products. Our operations could be interrupted if we encounter delays or difficulties in securing these reagents and enzymes, sequencers or other equipment or materials, and if we cannot obtain an acceptable substitute. Any such interruption could significantly affect our business, financial condition, results of operations and reputation. We believe that there are only a few other manufacturers that are currently capable of supplying and servicing the equipment necessary for our operations, including sequencers and various associated reagents and enzymes. The use of equipment or materials provided by these replacement suppliers would require us to alter our operations. Transitioning to a new supplier would be time consuming and expensive, may result in interruptions in operations, could affect the performance specifications of our laboratory operations or could require that we revalidate our tests. We cannot be certain that we will be able to secure alternative equipment, reagents and other materials, or bring such equipment, reagents and materials on line and revalidate them without experiencing interruptions in our workflow. If we encounter delays or difficulties in securing, reconfiguring or revalidating equipment and materials, our business and reputation could be adversely affected.
Customer concentration and seasonality
We receive payment for our products and services from patients, biopharmaceutical partners, third-party payers and other business-to-business customers. As of December 31, 2020, our revenue has been primarily derived from test reports generated from our assays. See information regarding our customer concentration in Note 2, “Summary of significant accounting policies” in Notes to Consolidated Financial Statements in Part II, Item 8 of this Annual Report.
We have historically experienced higher revenue in our fourth quarter compared to other quarters in our fiscal year due in part to seasonal demand of our tests from patients who have met their annual insurance deductible. However, changes in our product and payer mix might cause these historical seasonal patterns to be different than future patterns of revenue or financial performance.
Human capital resources
Our mission
The strength of our team and the culture in which we work is essential to our ability to achieve our broader mission. Attracting, developing and retaining exceptional employees is vitally important to us, but we also invest in creating a differentiated culture for our team that enables continuous innovation at scale. We want Invitae to be a force for good, a team that is helping make genetics and healthcare equally accessible to all. We had approximately 2,100 employees as of December 31, 2020, of which approximately 55% are women and 45% men.
23
Our employee engagement and culture
Our hiring process has been designed to provide an equitable candidate experience, facilitate the inclusion of new perspectives, foster innovation and creativity, and leverage technology and data analytics to address gaps in representation. In 2020 we established a Diversity, Equity and Inclusion, or DEI, roadmap. Our vision is to cultivate a place where we all belong. Our DEI mission is to engage, develop and retain talent from diverse backgrounds by fostering community, providing education and support, and advancing inclusive research and health equity globally. As of December 2020, excluding employees who joined us through our acquisition of ArcherDX in October 2020, approximately 59% of our workforce was White, 20% Asian, 8% Hispanic, 5% two or more races (not Hispanic or Latino), and 4% Black or African American.
We are committed to maintaining and improving the health and safety of our employees. As per our Code of Business Conduct and Ethics, all employees have responsibility for maintaining a safe and healthy workplace for all other employees by following our safety and health rules, policies and practices and reporting accidents, injuries and unsafe equipment, practices or conditions. In addition, we established a Crisis Management Team that, along with the Employee Health and Safety Administrator, comprise the Steering Committee for pandemic response.
We empower our employees to own their career path and seek out training programs to take them to the next level. We are currently in the process of developing a structure of growth opportunities and ways to understand and communicate pathways. We have also invested in our training and development programs and infrastructure for our employees.
As part of our commitment to data-driven decision making, we conduct an ongoing monthly survey that asks our teammates three key questions: how they feel about the company direction, what’s going well and what’s not going well. We’ve been asking these questions consistently for several years so we can quickly identify trends as they emerge. We believe this combination of ongoing pulse surveys to help detect timely changes in team morale and engagement, and occasional deep dives for a more complete picture, allows our management and Talent Operations to better understand team dynamics and make changes to policies, benefits and organizational structure to respond to current challenges. It allows us to quickly gather feedback on what’s working and what’s not.
Information about our Executive Officers
The names of our executive officers and other corporate officers, and their ages as of February 26, 2021, are as follows:
| Name | Age | Position | ||||||||||||
| Sean E. George, Ph.D. | 47 | President, Chief Executive Officer, Director and Co-Founder | ||||||||||||
| Thomas R. Brida | 50 | General Counsel and Secretary | ||||||||||||
| Shelly D. Guyer | 60 | Chief Financial Officer | ||||||||||||
| Kenneth D. Knight | 60 | Chief Operating Officer | ||||||||||||
| Robert L. Nussbaum, M.D. | 71 | Chief Medical Officer | ||||||||||||
| Katherine A. Stueland | 45 | Chief Commercial Officer | ||||||||||||
| Robert F. Werner | 47 | Chief Accounting Officer | ||||||||||||
Sean E. George, Ph.D. is one of our co-founders and has been our President and Chief Executive Officer since January 2017, a position he also held from January 2010 through August 2012. Dr. George also served as our President since August 2012 and he served as our Chief Operating Officer from August 2012 until January 2017. He has also served as a director since January 2010. Prior to co-founding Invitae, Dr. George served as Chief Operating Officer from 2007 to November 2009 at Navigenics, Inc., a personalized medicine company. Previously, he served as Senior Vice President of Marketing and Senior Vice President, Life Science Business at Affymetrix, Inc., a provider of life science and molecular diagnostic products, as well as Vice President, Labeling and Detection Business at Invitrogen Corporation, a provider of tools to the life sciences industry, during his tenure there from 2002 to 2007. Dr. George currently serves as a director of CM Life Sciences, Inc., a publicly traded special purpose acquisition company. Dr. George holds a B.S. in Microbiology and Molecular Genetics from the University of California Los Angeles, an M.S. in Molecular and Cellular Biology from the University of California Santa Barbara, and a Ph.D. in Molecular Genetics from the University of California Santa Cruz.
Thomas R. Brida has served as our General Counsel since January 2017. Mr. Brida also served as our Deputy General Counsel from January 2016 to January 2017. Prior to joining Invitae, he was Associate General Counsel at Bio-Rad Laboratories, a life science research and clinical diagnostics manufacturer, from January 2004 to January 2016. He holds a B.A. from Stanford University and a J.D. from the U.C. Berkeley School of Law.
24
Shelly D. Guyer has served as our Chief Financial Officer since June 2017. On November 5, 2020, we announced that Ms. Guyer will be transitioning to a new role leading our sustainability efforts, including our ESG (environmental, social and governance) initiatives. She will continue to serve as Chief Financial Officer while the Company conducts a search for her successor. Ms. Guyer served as Chief Financial Officer of Veracyte, Inc., a genomic diagnostics company, from April 2013 to December 2016 and served as Veracyte’s Secretary from April 2013 to March 2014. Previously, she served as Chief Financial Officer and Executive Vice President of Finance and Administration of iRhythm Technologies, Inc., a digital healthcare company, from April 2008 to December 2012. From March 2006 to August 2007, Ms. Guyer served as Vice President of Business Development and Investor Relations of Nuvelo, Inc., a biopharmaceutical company. Prior to joining Nuvelo, Ms. Guyer worked at J.P. Morgan Securities and its predecessor companies for over 17 years, serving in a variety of roles including in healthcare investment banking and four years with the H&Q Environmental Technology Fund. Ms. Guyer currently serves as a director and chair of the audit committee of NGM Biopharmaceuticals, Inc., a publicly held biopharmaceutical company. Ms. Guyer holds an A.B. in Politics from Princeton University and an M.B.A. from the Haas School of Business at the University of California Berkeley.
Kenneth D. Knight has served as our Chief Operating Officer since June 2020. Prior to that, he most recently served as Vice President of transportation services at Amazon.com, Inc., a multinational and diversified technology company, from December 2019 to June 2020, and as Vice President of Amazon’s global delivery services, fulfillment operations and human resources from April 2016 to December 2019. Prior to his time at Amazon, from 2012 to March 2016, Mr. Knight served as general manager of material handling and underground business division at Caterpillar Inc., a manufacturer of machinery and equipment. Prior to that, Mr. Knight served in various capacities at General Motors Company, a vehicle manufacturer, for 27 years, including as executive director of global manufacturing engineering and as manufacturing general manager. Mr. Knight holds a B.S. in Electrical Engineering from the Georgia Institute of Technology and a Master of Business Administration from the Massachusetts Institute of Technology.
Robert L. Nussbaum, M.D. has served as our Chief Medical Officer since August 2015. From April 2006 to August 2015, he was chief of the Division of Genomic Medicine at UCSF Health where he also held leadership roles in the Cancer Genetics and Prevention Program beginning in January 2009 and the Program in Cardiovascular Genetics beginning in July 2007. From April 2006 to August 2015, he served as a member of the UCSF Institute for Human Genetics. Prior to joining UCSF Health, Dr. Nussbaum was chief of the Genetic Disease Research Branch of the National Human Genome Research Institute, one of the National Institutes of Health, from 1994 to 2006. He is a member of the National Academy of Medicine and a fellow at the American Academy of Arts and Sciences. Dr. Nussbaum is a board-certified internist and medical geneticist who holds a B.S. in Applied Mathematics from Harvard College and an M.D. from Harvard Medical School in the Harvard-MIT joint program in Health Sciences and Technology. He completed his residency in internal medicine at Barnes-Jewish Hospital and a fellowship in medical genetics at the Baylor College of Medicine.
Katherine A. Stueland has served as our Chief Commercial Officer since October 2016. From January 2014 to October 2016, she served as our head of communications and investor relations. Prior to joining Invitae, Ms. Stueland was a Principal at Vivo Communications, a healthcare communications company, from January 2013 to December 2013. Previously, she served as Vice President, Communications and Investor Relations at Dendreon Corporation, a biotechnology company. Ms. Stueland holds a B.S in English Literature from Miami University in Ohio.
Robert F. Werner has served as our Chief Accounting and Principal Accounting Officer since May 2020. Prior to that, Mr. Werner served as our Corporate Controller from September 2017. Prior to joining Invitae, from February 2015 to September 2017, Mr. Werner served as Vice President of Finance and Corporate Controller of Proteus Digital Health, Inc., a digital medicine pharmaceuticals company. Prior to that, Mr. Werner served as Corporate Controller and Principal Accounting Officer of CardioDx, Inc., a molecular diagnostics company, from March 2012 to February 2015. Mr. Werner is a Certified Public Accountant and started his career at Ernst & Young LLP. Mr. Werner holds a Bachelor of Science in Accounting and a Master of Accountancy in Professional Accounting from Brigham Young University’s Marriott School of Management.
General Information
We were incorporated in the State of Delaware on January 13, 2010 under the name Locus Development, Inc. and changed our name to Invitae Corporation in 2012.
Our principal executive offices are located at 1400 16th Street, San Francisco, California 94103, and our telephone number is (415) 374-7782. Our website address is www.invitae.com. The information contained on, or that can be accessed through, our website is not part of this annual report on Form 10-K.
25
We make available free of charge on our website our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports, as soon as reasonably practicable after we electronically file or furnish such materials to the Securities and Exchange Commission, or SEC. You may obtain a free copy of these reports in the Investor Relations section of our website, www.invitae.com. All reports that we file are also available at www.sec.gov.
26
ITEM 1A. Risk Factors.
Risks related to our business and strategy
We face risks related to health epidemics, including the recent COVID-19 pandemic, which could have a material adverse effect on our business and results of operations.
Our business has been and could continue to be adversely affected by a widespread outbreak of contagious disease, including the recent pandemic of respiratory illness caused by a novel coronavirus known as COVID-19. Global health concerns relating to COVID-19 have negatively affected the macroeconomic environment, and the pandemic has significantly increased economic volatility and uncertainty.
The pandemic has resulted in government authorities implementing numerous measures to try to contain the virus, such as travel bans and restrictions, quarantines, shelter-in-place or stay-at-home orders, and business shutdowns. For example, some of our personnel located at our headquarters and other offices in California, elsewhere in the United States and in other countries, have been subject to shelter-in-place or stay-at-home orders from state and local governments. These measures have adversely impacted and may further impact our employees and operations and the operations of our customers, suppliers and business partners, and may continue to negatively impact spending patterns, payment cycles and insurance coverage levels. These measures have adversely affected and may continue to adversely affect demand for our tests. Earlier this year, many of our customers, including hospitals and clinics, suspended non-emergency appointments and services, which resulted in a significant decrease in our test volume. Travel bans, restrictions and border closures have also impacted our ability to ship tests to and receive samples from our customers. Some of these measures by government authorities have and may continue to remain in place for a significant period of time. Even if these measures are lifted, they may be implemented again if COVID-19 is not contained or returns, as has been the case recently. These measures have adversely affected and may continue to adversely affect our test volume, sales activities and results of operations.
The spread of COVID-19 has caused us to modify our business practices (including employee travel, mandating that all non-essential personnel work from home, temporary closures of our offices, cancellation of physical participation in sales activities, meetings, events and conferences and increasing inventories of certain supplies because, although we have not experienced significant disruption in our supply chain, over the past several months, we have experienced supply delays as a result of the COVID-19 pandemic and have also had to obtain supplies from new suppliers), and we may take further actions as may be required by government authorities or that we determine are in the best interests of our employees, customers and business partners. Such actions could also impact our ability to fully integrate businesses we have acquired and those we may acquire in the future. There is no certainty that such actions will be sufficient to mitigate the risks posed by the virus or otherwise be satisfactory to government authorities. If significant portions of our workforce are unable to work effectively, including due to illness, quarantines, social distancing, government actions or other restrictions in connection with COVID-19, our operations will be impacted.
The extent to which COVID-19 continues to impact our business, results of operations and financial condition will depend on future developments, which are highly uncertain and cannot be predicted, including, but not limited to, the duration and spread of the pandemic, the actions to contain the virus and treat its impact, and how quickly and to what extent normal economic and operating activities can resume. COVID-19 could limit the ability of our customers, suppliers and business partners to perform, including third-party payers’ ability to make timely payments to us during and following the pandemic. We have also experienced and may continue to experience a shortage of, or delays in, laboratory supplies and equipment, or a suspension of services from other laboratories or third parties. Even after COVID-19 has subsided, we may continue to experience an adverse impact to our business as a result of its global economic impact, including any recession that has occurred or may occur in the future, and loss of health insurance coverage resulting from pandemic-related job losses.
Specifically, difficult macroeconomic conditions, such as decreases in per capita income and level of disposable income, increased and prolonged unemployment or a decline in consumer confidence as a result of COVID-19, as well as limited or significantly reduced points of access of our tests, could have a material adverse effect on the demand for our tests. Under difficult economic conditions, consumers may seek to reduce discretionary spending by forgoing our tests. Decreased demand for our tests, particularly in the United States, has negatively affected and could continue to negatively affect our overall financial performance. Because a significant portion of our revenue is concentrated in the United States, where the impact of COVID-19 has been significant, COVID-19 has had and could continue to have a disproportionately negative impact on our business and financial results.
27
There are no comparable recent events which may provide guidance as to the effect of the spread of COVID-19 and a pandemic, and, as a result, the ultimate impact of COVID-19 or a similar health epidemic is highly uncertain and subject to change. We do not yet know the full extent of COVID-19’s impact on our business, our operations, or the global economy as a whole. However, the effects could have a material impact on our results of operations, and we continue to monitor the situation closely.
To the extent the COVID-19 pandemic continues to adversely affect our business and financial results, it may also have the effect of heightening many of the other risks described in this section.
We expect to continue incurring significant losses, and we may not successfully execute our plan to achieve or sustain profitability.
We have incurred substantial losses since our inception. For the years ended December 31, 2020, 2019 and 2018, our net losses were $602.2 million, $242.0 million and $129.4 million, respectively. At December 31, 2020, our accumulated deficit was $1.4 billion. While our revenue has increased over time, we expect to continue to incur significant losses as we invest in our business. We incurred research and development expenses of $240.6 million, $141.5 million and $63.5 million in 2020, 2019 and 2018, respectively, and selling and marketing expenses of $168.3 million, $122.2 million and $74.4 million in 2020, 2019 and 2018, respectively. We expect these losses may increase as we focus on scaling our business and operations and expanding our testing capabilities, which may also increase our operating expenses, and we have experienced and may continue to experience decreases in test volume due to the impact of COVID-19. In addition, as a result of the integration of acquired businesses, we may be subject to unforeseen or additional expenditures, costs or liabilities, including costs and potential liabilities associated with litigation. Our prior losses and expected future losses have had and may continue to have an adverse effect on our stockholders’ equity, working capital and stock price. Our failure to achieve and sustain profitability in the future would negatively affect our business, financial condition, results of operations and cash flows, and could cause the market price of our common stock to decline.
We began operations in January 2010 and commercially launched our initial assay in late November 2013; accordingly, we have a relatively limited operating history upon which you can evaluate our business and prospects. Our limited commercial history makes it difficult to evaluate our current business and makes predictions about our future results, prospects or viability subject to significant uncertainty. Our prospects must be considered in light of the risks and difficulties frequently encountered by companies in their early stage of development, particularly companies in new and rapidly evolving markets such as ours. These risks include an evolving and unpredictable business model and the management of growth. To address these risks, we must, among other things, increase our customer base; continue to implement and successfully execute our business and marketing strategy; identify, acquire and successfully integrate companies, assets or technologies in areas that are complementary to our business strategy; successfully enter into other strategic collaborations or relationships; obtain access to capital on acceptable terms and effectively utilize that capital; identify, attract, hire, retain, motivate and successfully integrate additional employees; continue to expand, automate and upgrade our laboratory, technology and data systems; obtain, maintain and expand coverage and reimbursement by healthcare payers; provide rapid test turnaround times with accurate results at low prices; provide superior customer service; and respond to competitive developments. We cannot assure you that we will be successful in addressing these risks, and the failure to do so could have a material adverse effect on our business, prospects, financial condition and results of operations.
Our inability to raise additional capital on acceptable terms in the future may limit our ability to develop and commercialize new tests and expand our operations.
We expect capital expenditures and operating expenses to increase over the next several years as we expand our infrastructure, commercial operations, research and development and selling and marketing activities and pursue and integrate acquisitions. We believe our existing cash and cash equivalents as of December 31, 2020, including the net proceeds from our recent public offering and revenue from sales of our tests will be sufficient to meet our anticipated cash requirements for our currently-planned operations for the foreseeable future. We may raise additional capital to finance operations prior to achieving profitability, or should we make additional acquisitions. We may seek to raise additional capital through equity offerings, debt financings, collaborations or licensing arrangements. Additional funding may not be available to us on acceptable terms, or at all. In addition, the terms of our credit agreement restrict our ability to incur certain indebtedness and issue certain equity securities. If we raise funds by issuing equity securities, dilution to our stockholders would result. Any equity securities issued also may provide for rights, preferences or privileges senior to those of holders of our common stock. The terms of debt securities issued or borrowings, if available, could impose significant restrictions on our operations.
28
The incurrence of additional indebtedness or the issuance of certain equity securities could result in increased fixed payment obligations and could also result in restrictive covenants, such as limitations on our ability to incur additional debt or issue additional equity, limitations on our ability to acquire or license intellectual property rights, and other operating restrictions that could adversely affect our ability to conduct our business. In addition, the issuance of additional equity securities by us, or the possibility of such issuance, may cause the market price of our common stock to decline. In the event we enter into collaborations or licensing arrangements to raise capital, we may be required to accept unfavorable terms. These agreements may require that we relinquish or license to a third party on unfavorable terms our rights to tests we otherwise would seek to develop or commercialize ourselves, or reserve certain opportunities for future potential arrangements when we might be able to achieve more favorable terms. If we are not able to secure additional funding when needed, we may have to delay, reduce the scope of or eliminate one or more research and development programs, selling and marketing initiatives, or potential acquisitions. In addition, we may have to work with a partner on one or more aspects of our tests or market development programs, which could lower the economic value of those tests or programs to our company.
We have acquired and may continue to acquire businesses or assets, form joint ventures or make investments in other companies or technologies that could harm our operating results, dilute our stockholders’ ownership, or cause us to incur debt or significant expense.
As part of our business strategy, we have pursued and expect to continue to pursue acquisitions of complementary businesses or assets, as well as technology licensing arrangements. We also may pursue strategic alliances that leverage our core technology and industry experience to expand our offerings or distribution, or make investments in other companies. Since 2017, we have acquired several companies, including companies in family health genetic information services, the patient data collection industry, the non-invasive prenatal screen offering industry, the genetic information industry and the use of artificial intelligence in such industry, the pharmacogenetic testing industry, and the oncology industry.
With respect to our acquired businesses and any acquisitions we may make in the future, we may not be able to integrate these acquisitions successfully into our existing business, and we could assume unknown or contingent liabilities. For example, if we are unable to integrate ArcherDX's technology, people and distributed products business model into our existing business, we will not realize the expected benefits of that acquisition. Any acquisitions by us also could result in significant write-offs or the incurrence of debt and contingent liabilities, any of which could harm our operating results. Furthermore, as we experienced in the past, the loss of customers, payers, partners or suppliers following the completion of any acquisitions by us could harm our business. Changes in services, sources of revenue, and branding or rebranding initiatives may involve substantial costs and may not be favorably received by customers, resulting in an adverse impact on our financial results, financial condition and stock price. Integration of an acquired company or business also may require management’s time and resources that otherwise would be available for ongoing development of our existing business. We may also need to divert cash from other uses to fund these integration activities and these new businesses. Ultimately, we may not realize the anticipated benefits of any acquisition, technology license, strategic alliance, joint venture or investment, or these benefits may take longer to realize than we expected.
In connection with certain of our completed acquisitions, we have agreed to pay cash and/or stock consideration that is contingent upon the achievement of specified objectives, such as development objectives, regulatory submissions, regulatory approvals and revenue recognized related to certain products. As of the date of the applicable acquisition, we record a contingent liability representing the estimated fair value of the contingent consideration we expect to pay. On a quarterly basis, we reassess these obligations and, in the event our estimate of the fair value of the contingent consideration changes, we recorded increases or decreases in the fair value as an adjustment to operating expense, which could have a material impact on our results of operations. As of December 31, 2020, we accrued $796.6 million of contingent consideration, most of which related to potential milestone payments in the form of our common stock in connection with our acquisition of ArcherDX. In addition, our actual payments may differ materially from the amount of the contingent liability, which could have a material impact on our results of operations.
To finance any acquisitions or investments, we may raise additional funds, which could adversely affect our existing stockholders and our business, as discussed in the preceding risk factor. If the price of our common stock is low or volatile, we may not be able to acquire other companies for stock. In addition, our stockholders may experience substantial dilution as a result of additional securities we may issue for acquisitions. Open market sales of substantial amounts of our common stock issued to stockholders of companies we acquire could also depress our stock price. Additional funds may not be available on terms that are favorable to us, or at all.
29
If third-party payers, including managed care organizations, private health insurers and government health plans do not provide adequate reimbursement for our tests or we are unable to comply with their requirements for reimbursement, our commercial success could be negatively affected.
Our ability to increase the number of billable tests and our revenue will depend on our success achieving reimbursement for our tests from third-party payers. Reimbursement by a payer may depend on a number of factors, including a payer’s determination that a test is appropriate, medically necessary, cost-effective and has received prior authorization. The commercial success of our distributed products, including STRATAFIDE, a pan-solid tumor in vitro diagnostic, or IVD, and our Personalized Cancer Monitoring product, or PCM, if approved, will depend on the extent to which our customers receive coverage and adequate reimbursement from third-party payers, including as managed care organizations and government payers (e.g., Medicare and Medicaid).
Since each payer makes its own decision as to whether to establish a policy or enter into a contract to cover our tests, as well as the amount it will reimburse for a test, seeking these approvals is a time-consuming and costly process. In addition, the determination by a payer to cover and the amount it will reimburse for our tests will likely be made on an indication by indication basis. To date, we have obtained policy-level reimbursement approval or contractual reimbursement for some indications for our tests from most of the large commercial third-party payers in the United States, and the Centers for Medicare & Medicaid Services, or CMS, provides reimbursement for our multi-gene tests for hereditary breast and ovarian cancer-related disorders as well as colon cancer. We believe that establishing adequate reimbursement from Medicare is an important factor in gaining adoption from healthcare providers. Our claims for reimbursement from third-party payers may be denied upon submission, and we must appeal the claims. The appeals process is time consuming and expensive and may not result in payment. In cases where there is not a contracted rate for reimbursement, there is typically a greater coinsurance or copayment requirement from the patient, which may result in further delay or decreased likelihood of collection.
In cases where we have established reimbursement rates with third-party payers, we face additional challenges in complying with their procedural requirements for reimbursement. These requirements often vary from payer to payer, and we have needed additional time and resources to comply with them. We have also experienced, and may continue to experience, delays in or denials of coverage if we do not adequately comply with these requirements. Our third-party payers have also requested, and in the future may request, audits of the amounts paid to us. We have been required to repay certain amounts to payers as a result of such audits, and we could be adversely affected if we are required to repay other payers for alleged overpayments due to lack of compliance with their reimbursement policies. In addition, we have experienced, and may continue to experience, delays in reimbursement when we transition to being an in-network provider with a payer.
We expect to continue to focus our resources on increasing adoption of, and expanding coverage and reimbursement for, our current tests and any future tests we may develop or acquire. If we fail to expand and maintain broad adoption of, and coverage and reimbursement for, our tests, our ability to generate revenue could be harmed and our future prospects and our business could suffer.
We face intense competition, which is likely to intensify further as existing competitors devote additional resources to, and new participants enter, the market. If we cannot compete successfully, we may be unable to increase our revenue or achieve and sustain profitability.
With the development of next generation sequencing, the clinical genetics market is becoming increasingly competitive, and we expect this competition to intensify in the future. We face competition from a variety of sources, including:
•dozens of relatively specialized competitors focused on genetics applied to healthcare, such as Ambry Genetics, a subsidiary of Konica Minolta Inc.; Athena Diagnostics and Blueprint Genetics, subsidiaries of Quest Diagnostics Incorporated; Baylor-Miraca Genetics Laboratories; Caris Life Sciences, Inc.; Centogene AG; Connective Tissue Gene Test LLC, a subsidiary of Health Network Laboratories, L.P.; Cooper Surgical, Inc.; Emory Genetics Laboratory, a subsidiary of Eurofins Scientific; Foundation Medicine, a subsidiary of Roche Holding AG; Fulgent Genetics, Inc., GeneDx, a subsidiary of OPKO Health, Inc.; Guardant Health, Inc.; Integrated Genetics, Sequenom Inc., Correlagen Diagnostics, Inc., and MNG Laboratories, subsidiaries of Laboratory Corporation of America Holdings; Myriad Genetics, Inc.; Natera, Inc.; Perkin Elmer, Inc.; PreventionGenetics, LLC; Progenity, Inc.; and Sema4 Genomics;
•a few large, established general testing companies with large market share and significant channel power, such as Laboratory Corporation of America Holdings and Quest Diagnostics Incorporated;
•a large number of clinical laboratories in an academic or healthcare provider setting that perform clinical genetic testing on behalf of their affiliated institutions and often sell and market more broadly; and
30
•a large number of new entrants into the market for genetic information ranging from informatics and analysis pipeline developers to focused, integrated providers of genetic tools and services for health and wellness including Illumina, Inc., which is also one of our suppliers.
Hospitals, academic medical centers and eventually physician practice groups and individual clinicians may also seek to perform at their own facilities the type of genetic testing we would otherwise perform for them. In this regard, continued development of equipment, reagents, and other materials as well as databases and interpretation services may enable broader direct participation in genetic testing and analysis.
Participants in closely related markets such as clinical trial or companion diagnostic testing could converge on offerings that are competitive with the type of tests we perform. Instances where potential competitors are aligned with key suppliers or are themselves suppliers could provide such potential competitors with significant advantages.
In addition, the biotechnology and genetic testing fields are intensely competitive both in terms of service and price, and continue to undergo significant consolidation, permitting larger clinical laboratory service providers to increase cost efficiencies and service levels, resulting in more intense competition.
We also face competition as a result of our recently completed acquisition of ArcherDX. In particular, ArcherDX competes with numerous companies in the life sciences research, clinical diagnostics and drug development spaces, including, among others, Natera, QIAGEN N.V., Guardant Health, Inc., Thermo Fisher, Inc., Foundation Medicine, Caris Life Sciences, Inc., Tempus, Laboratory Corporation of America, Quest Diagnostics, Inc., NeoGenomics, Inc., BioReference Laboratories, Inc. and Illumina, Inc.
We believe the principal competitive factors in our market are:
•breadth and depth of content;
•quality;
•reliability;
•accessibility of results;
•turnaround time of testing results;
•price and quality of tests;
•coverage and reimbursement arrangements with third-party payers;
•convenience of testing;
•brand recognition of test provider;
•additional value-added services and informatics tools;
•client service; and
•quality of website content.
31
Many of our competitors and potential competitors have longer operating histories, larger customer bases, greater brand recognition and market penetration, higher margins on their tests, substantially greater financial, technological and research and development resources, selling and marketing capabilities, lobbying efforts, and more experience dealing with third-party payers. As a result, they may be able to respond more quickly to changes in customer requirements, devote greater resources to the development, promotion and sale of their tests than we do, sell their tests at prices designed to win significant levels of market share, or obtain reimbursement from more third-party payers and at higher prices than we do. We may not be able to compete effectively against these organizations. Increased competition and cost-saving initiatives on the part of governmental entities and other third-party payers are likely to result in pricing pressures, which could harm our sales, profitability or ability to gain market share. In addition, competitors may be acquired by, receive investments from or enter into other commercial relationships with larger, well-established and well-financed companies as use of next generation sequencing for clinical diagnosis and preventative care increases. Certain of our competitors may be able to secure key inputs from vendors on more favorable terms, devote greater resources to marketing and promotional campaigns, adopt more aggressive pricing policies and devote substantially more resources to website and systems development than we can. In addition, companies or governments that control access to genetic testing through umbrella contracts or regional preferences could promote our competitors or prevent us from performing certain services. In addition, some of our competitors have obtained approval or clearance for certain of their tests from the U.S. Food and Drug Administration, or FDA. If payers decide to reimburse only for tests that are FDA-approved or FDA-cleared, or if they are more likely to reimburse for such tests, we may not be able to compete effectively unless we obtain similar approval or clearance for our tests. If we are unable to compete successfully against current and future competitors, we may be unable to increase market acceptance and sales of our tests, which could prevent us from increasing our revenue or achieving profitability and could cause our stock price to decline.
We may not be able to manage our future growth effectively, which could make it difficult to execute our business strategy.
Our expected future growth could create a strain on our organizational, administrative and operational infrastructure, including laboratory operations, quality control, customer service, marketing and sales, and management. We may not be able to maintain the quality of or expected turnaround times for our tests, or satisfy customer demand as it grows. We may need to continue expanding our sales force to facilitate our growth, and we may have difficulties locating, recruiting, training and retaining sales personnel. Our ability to manage our growth effectively will require us to continue to improve our operational, financial and management controls, as well as our reporting systems and procedures. As we grow, any failure of our controls or interruption of our production facilities or systems could have a negative impact on our business and financial operations. We plan to implement new enterprise software systems in a number of areas affecting a broad range of business processes and functional areas. The time and resources required to implement these new systems is uncertain, and failure to complete these activities in a timely and efficient manner could adversely affect our operations. Future growth in our business could also make it difficult for us to maintain our corporate culture. If we are unable to manage our growth effectively, it may be difficult for us to execute our business strategy and our business could be harmed.
32
Security breaches, privacy issues, loss of data and other incidents could compromise sensitive or personal information related to our business or prevent us from accessing critical information and expose us to liability, which could adversely affect our business and our reputation.
In the ordinary course of our business, we collect and store sensitive data, including protected health information, or PHI, personally identifiable information, genetic information, credit card information, intellectual property and proprietary business information owned or controlled by ourselves or our customers, payers and other parties. We manage and maintain our applications and data utilizing a combination of on-site systems, managed data center systems and cloud-based systems. We also communicate PHI and other sensitive patient data through our various customer tools and platforms. In addition to storing and transmitting sensitive data that is subject to multiple legal protections, these applications and data encompass a wide variety of business-critical information including research and development information, commercial information, and business and financial information. We face a number of risks relative to protecting this critical information, including loss of access risk, inappropriate disclosure, inappropriate modification, and the risk of our being unable to adequately monitor and modify our controls over our critical information. Any technical problems that may arise in connection with our data and systems, including those that are hosted by third-party providers, could result in interruptions to our business and operations or exposure to security vulnerabilities. These types of problems may be caused by a variety of factors, including infrastructure changes, intentional or accidental human actions or omissions, software errors, malware, viruses, security attacks, fraud, spikes in customer usage and denial of service issues. From time to time, large third-party web hosting providers have experienced outages or other problems that have resulted in their systems being offline and inaccessible. Such outages could materially impact our business and operations.
The secure processing, storage, maintenance and transmission of this critical information are vital to our operations and business strategy, and we devote significant resources to protecting such information. Although we take what we believe to be reasonable and appropriate measures, including a formal, dedicated enterprise security program, to protect sensitive information from various compromises (including unauthorized access, disclosure, or modification or lack of availability), our information technology and infrastructure may be vulnerable to attacks by hackers or viruses or breached due to employee error, malfeasance or other disruptions. For example, ArcherDX has been subject to phishing incidents and we may experience additional incidents in the future. Any such breach or interruption could compromise our networks and the information stored therein could be accessed by unauthorized parties, altered, publicly disclosed, lost or stolen.
Further, our various customer tools and platforms are currently accessible through our online portal and/or through our mobile applications, and there is no guarantee we can protect our them from a security breach. Unauthorized access, loss or dissemination could also disrupt our operations (including our ability to conduct our analyses, provide test results, bill payers or patients, process claims and appeals, provide customer assistance, conduct research and development activities, collect, process and prepare company financial information, provide information about our tests and other patient and physician education and outreach efforts through our website, and manage the administrative aspects of our business) and damage our reputation, any of which could adversely affect our business.
In addition to data security risks, we also face privacy risks. Should we actually violate, or be perceived to have violated, any privacy promises we make to patients or consumers, we could be subject to a complaint from an affected individual or interested privacy regulator, such as the FTC, a state Attorney General, an EU Member State Data Protection Authority, or a data protection authority in another international jurisdiction. This risk is heightened given the sensitivity of the data we collect.
Any security compromise that causes an apparent privacy violation could also result in legal claims or proceedings; liability under federal, state, foreign, or multinational laws that regulate the privacy, security, or breach of personal information, such as but not limited to the HIPAA, HITECH, state data security and data breach notification laws, the European Union’s General Data Protection Regulation, or GDPR, the UK Data Protection Act of 2018; and related regulatory penalties. Penalties for failure to comply with a requirement of HIPAA or HITECH vary significantly, and, depending on the knowledge and culpability of the HIPAA-regulated entity, may include civil monetary penalties of up to $1.5 million per calendar year for each provision of HIPAA that is violated. A person who knowingly obtains or discloses individually identifiable health information in violation of HIPAA may face a criminal penalty of up to $50,000 and up to one-year imprisonment. The criminal penalties increase if the wrongful conduct involves false pretenses or the intent to sell, transfer or use identifiable health information for commercial advantage, personal gain or malicious harm. Penalties for unfair or deceptive acts or practices under the FTC Act or state Unfair and Deceptive Acts and Practices, or UDAP, statutes may also vary significantly.
33
There has been unprecedented activity in the development of data protection regulation around the world. As a result, the interpretation and application of consumer, health-related and data protection laws in the United States, Europe and elsewhere are often uncertain, contradictory and in flux. The GDPR took effect on May 25, 2018. The GDPR applies to any entity established in the EU as well as extraterritorially to any entity outside the EU that offers goods or services to, or monitors the behavior of, individuals who are located in the EU. The GDPR imposes strict requirements on controllers and processors of personal data, including enhanced protections for “special categories” of personal data, which includes sensitive information such as health and genetic information of data subjects. The GDPR also grants individuals various rights in relation to their personal data, including the rights of access, rectification, objection to certain processing and deletion. The GDPR provides an individual with an express right to seek legal remedies if the individual believes his or her rights have been violated. Failure to comply with the requirements of the GDPR or the related national data protection laws of the member states of the EU, which may deviate from or be more restrictive than the GDPR, may result in significant administrative fines issued by EU regulators. Maximum penalties for violations of the GDPR are capped at 20M euros or 4% of an organization’s annual global revenue, whichever is greater.
Further, the United Kingdom’s decision to leave the European Union, often referred to as Brexit, has created uncertainty with regard to data protection regulation in the United Kingdom. In particular, it is still unclear whether the transfer of personal information from the EU to the United Kingdom will in the future remain lawful under the GDPR. The United Kingdom-EU post-Brexit trade deal provides that transfers of personal information to the United Kingdom will not be treated as restricted transfers to a non-EU country for a period of up to six months from January 1, 2021. However, unless the EU Commission makes an “adequacy finding” with respect to the United Kingdom before the end of that transition period, from that date the United Kingdom will be a “third country” under the GDPR and transfers of personal information from the EU to the United Kingdom will require an “adequacy mechanism,” such as the SCCs.
Additionally, the implementation of GDPR has led other jurisdictions to either amend or propose legislation to amend their existing data privacy and cybersecurity laws to resemble the requirements of GDPR. For example, on June 28, 2018, California adopted the CCPA. The CCPA regulates how certain for-profit businesses that meet one or more CCPA applicability thresholds collect, use, and disclose the personal information of consumers who reside in California. Among other things, the CCPA confers to California consumers the right to receive notice of the categories of personal information that will be collected by a business, how the business will use and share the personal information, and the third parties who will receive the personal information; the CCPA also confers rights to access, delete, or transfer personal information; and the right to receive equal service and pricing from a business after exercising a consumer right granted by the CCPA. In addition, the CCPA allows California consumers the right to opt out of the “sale” of their personal information, which the CCPA defines broadly as any disclosure of personal information to a third party in exchange for monetary or other valuable consideration. The CCPA also requires a business to implement reasonable security procedures to safeguard personal information against unauthorized access, use, or disclosure. California amended the law in September 2018 to exempt all PHI collected by certain parties subject to HIPAA, and further amended the law in September 2020 to clarify that de-identified data as defined under HIPAA will also be exempt from the CCPA. The California Attorney General’s final regulations implementing the CCPA took effect on August 14, 2020. The CCPA provides for civil penalties for violations, as well as a private right of action for data breaches resulting from a business’s failure to implement and maintain reasonable data security procedures that is expected to increase data breach litigation.
In addition, California voters recently approved the California Privacy Rights Act of 2020, or CPRA, that is scheduled to go into effect on January 1, 2023. The CPRA would, among other things, amend the CCPA to give California residents the ability to limit the use of their sensitive information, provide for penalties for CPRA violations concerning California residents under the age of 16, and establish a new California Privacy Protection Agency to implement and enforce the law. Other jurisdictions in the United States are beginning to propose laws similar to the CCPA. Some observers have noted that the CCPA could mark the beginning of a trend toward more stringent privacy legislation in the United States, which could increase our potential liability and adversely affect our business, results of operations, and financial condition.
34
It is possible the GDPR, CCPA and other emerging United States and international data protection laws may be interpreted and applied in a manner that is inconsistent with our practices. If so, this could result in government-imposed fines or orders requiring that we change our practices, which could adversely affect our business. In addition, these privacy laws and regulations may differ from country to country and state to state, and our obligations under these laws and regulations vary based on the nature of our activities in the particular jurisdiction, such as whether we collect samples from individuals in the local jurisdiction, perform testing in the local jurisdiction, or process personal information regarding employees or other individuals in the local jurisdiction. Complying with these various laws and regulations could cause us to incur substantial costs or require us to change our business practices and compliance procedures in a manner adverse to our business. We can provide no assurance that we are or will remain in compliance with diverse privacy and data security requirements in all of the jurisdictions in which we do business. Failure to comply with privacy and data security requirements could result in a variety of consequences, including civil or criminal penalties, litigation, or damage to our reputation, any of which could have a material adverse effect on our business.
We rely on highly skilled personnel in a broad array of disciplines and, if we are unable to hire, retain or motivate these individuals, or maintain our corporate culture, we may not be able to maintain the quality of our services or grow effectively.
Our performance, including our research and development programs and laboratory operations, largely depend on our continuing ability to identify, hire, develop, motivate and retain highly skilled personnel for all areas of our organization, including software developers, geneticists, biostatisticians, certified laboratory scientists and other scientific and technical personnel to process and interpret our genetic tests. In addition, we may need to continue to expand our sales force with qualified and experienced personnel. Competition in our industry for qualified employees is intense, and we may not be able to attract or retain qualified personnel in the future due to the competition for qualified personnel among life science and technology businesses as well as universities and public and private research institutions, particularly in the San Francisco Bay Area. In addition, our compensation arrangements, such as our equity award programs, may not always be successful in attracting new employees and retaining and motivating our existing employees. If we are not able to attract and retain the necessary personnel to accomplish our business objectives, we may experience constraints that could adversely affect our ability to scale our business, support our research and development efforts and our clinical laboratory. We believe that our corporate culture fosters innovation, creativity and teamwork. However, as our organization grows, we may find it increasingly difficult to maintain the beneficial aspects of our corporate culture. This could negatively impact our ability to retain and attract employees and our future success.
We need to scale our infrastructure in advance of demand for our tests, and our failure to generate sufficient demand for our tests would have a negative impact on our business and our ability to attain profitability.
Our success depends in large part on our ability to extend our market position, to provide customers with high-quality test reports quickly and at a lower price than our competitors, and to achieve sufficient test volume to realize economies of scale. In order to execute our business model, we intend to continue to invest heavily in order to significantly scale our infrastructure, including our testing capacity and information systems, expand our commercial operations, customer service, billing and systems processes and enhance our internal quality assurance program. We expect that much of this growth will be in advance of demand for our tests. Our current and future expense levels are to a large extent fixed and are largely based on our investment plans and our estimates of future revenue. Because the timing and amount of revenue from our tests is difficult to forecast, when revenue does not meet our expectations, we may not be able to adjust our spending promptly or reduce our spending to levels commensurate with our revenue. Even if we are able to successfully scale our infrastructure and operations, we cannot assure you that demand for our tests will increase at levels consistent with the growth of our infrastructure. If we fail to generate demand commensurate with this growth or if we fail to scale our infrastructure sufficiently in advance of demand to successfully meet such demand, our business, prospects, financial condition and results of operations could be adversely affected.
35
If we are not able to continue to generate substantial demand of our tests, our commercial success will be negatively affected.
Our business model assumes that we will be able to generate significant test volume, and we may not succeed in continuing to drive adoption of our tests to achieve sufficient volumes. Inasmuch as detailed genetic data from broad-based testing panels such as our tests have only recently become available at relatively affordable prices, the continued pace and degree of clinical acceptance of the utility of such testing is uncertain. Specifically, it is uncertain how much genetic data will be accepted as necessary or useful, as well as how detailed that data should be, particularly since medical practitioners may have become accustomed to genetic testing that is specific to one or a few genes. Given the substantial amount of additional information available from a broad-based testing panel such as ours, there may be distrust as to the reliability of such information when compared with more limited and focused genetic tests. To generate further demand for our tests, we will need to continue to make clinicians aware of the benefits of our tests, including the price, the breadth of our testing options, and the benefits of having additional genetic data available from which to make treatment decisions. A lack of or delay in clinical acceptance of broad-based panels such as our tests would negatively impact sales and market acceptance of our tests and limit our revenue growth and potential profitability. Genetic testing is expensive and many potential customers may be sensitive to pricing. In addition, potential customers may not adopt our tests if adequate reimbursement is not available, or if we are not able to maintain low prices relative to our competitors. Also, we may not be successful in increasing demand for our tests through our direct channel, in which we facilitate the ordering of our genetic tests by consumers through an online network of physicians.
If we are not able to generate demand for our tests at sufficient volume, or if it takes significantly more time to generate this demand than we anticipate, our business, prospects, financial condition and results of operations could be materially harmed.
We have devoted a portion of our resources to the development and commercialization of STRATAFIDE, and to research and development activities related to our PCM product for cancer monitoring, including clinical and regulatory initiatives to obtain diagnostic clearance and marketing approval. The demand for these regulated products is unproven, and we may not be successful in achieving market awareness and demand for these products through our sales and marketing operations.
If ArcherDX’s products and services do not perform as expected, we may not realize the expected benefits of our recent acquisition of ArcherDX.
The success of ArcherDX’s products depends on the market’s confidence that it can provide reliable products that enable high quality diagnostic testing with high sensitivity and specificity and short turnaround times. There is no guarantee that the accuracy and reproducibility ArcherDX has demonstrated to date will continue as its product deliveries increase and its product portfolio expands.
ArcherDX’s products and services use a number of complex and sophisticated biochemical and bioinformatics processes, many of which are highly sensitive to external factors. An operational, technological or other failure in one of these complex processes or fluctuations in external variables may result in sensitivity or specificity rates that are lower than ArcherDX anticipates or result in longer than expected turnaround times. In addition, labs are required to validate their processes before using ArcherDX products for clinical purposes. These validations are outside of our control. If our products do not perform, or are perceived to not have performed, as expected or favorably to competitive products, our consolidated operating results, reputation, and business will suffer, and ArcherDX may also be subject to legal claims arising from product limitations, errors, or inaccuracies.
In addition, we plan to match our test reports for STRATAFIDE to identified mutations with FDA-approved targeted therapies or relevant clinical trials of targeted therapies. If a patient or physician who orders a test using one of our products is unable to obtain, or be reimbursed for the use of, targeted therapies because they are not indicated in the FDA-approved label for treatment, the patient is unable to enroll in an identified clinical trial due to the enrollment criteria of the trial, or some other reason, the ordering physician may conclude the test report does not contain actionable information. If physicians do not believe our products consistently generate actionable information about their patients’ disease or condition, they may be less likely to use our products.
Furthermore, we cannot provide assurance that customers will always use these products in the manner in which intended. Any intentional or unintentional misuse of these products by customers could lead to substantial civil and criminal monetary and non-monetary penalties, and could result in significant legal and investigatory fees.
36
The future growth of our distributed products business is partially dependent upon regulatory approval and market acceptance of our IVD products, including STRATAFIDE and PCM.
We anticipate that the future success of our distributed products business will depend in large part on our ability to effectively introduce enhanced or new offerings of IVD products, such as STRATAFIDE. The development and launch of enhanced or new products and services, whether research use only, or RUO, or IVD, require the completion of certain clinical development and commercialization activities that are complex, costly, time-intensive and uncertain, and require us to accurately anticipate patients’, providers’ and, if applicable, payers’ attitudes and needs and emerging technology and industry trends. This process is conducted in various stages, and each stage presents the risk that ArcherDX will not achieve its goals on a timely basis, or at all.
We have limited experience commercializing IVD products. We may experience research and development, regulatory, marketing and other difficulties that could delay or prevent our introduction of enhanced or new products or new services and result in increased costs and the diversion of management’s attention and resources from other business matters.
An important factor in our ability to commercialize our distributed products is collecting data that supports their value proposition. The data collected from any studies we complete may not be favorable or consistent with its existing data or may not be statistically significant or compelling to the medical community or to third-party payers seeking such data for purposes of determining coverage for these products. This is particularly true with respect to service defects and errors. Any of the foregoing could have a negative impact on our ability to commercialize our future products, which could have a material adverse effect on our ability to realize the intended benefits of our recent acquisition of ArcherDX.
Our success will depend on our ability to use rapidly changing genetic data to interpret test results accurately and consistently, and our failure to do so would have an adverse effect on our operating results and business, harm our reputation and could result in substantial liabilities that exceed our resources.
Our success depends on our ability to provide reliable, high-quality tests that incorporate rapidly evolving information about the role of genes and gene variants in disease and clinically relevant outcomes associated with those variants. Errors, such as failure to detect genomic variants with high accuracy, or mistakes, such as failure to identify, or incompletely or incorrectly identifying, gene variants or their significance, could have a significant adverse impact on our business.
Hundreds of genes can be implicated in some disorders, and overlapping networks of genes and symptoms can be implicated in multiple conditions. As a result, a substantial amount of judgment is required in order to interpret testing results for an individual patient and to develop an appropriate patient report. We classify variants in accordance with published guidelines as benign, likely benign, variants of uncertain significance, likely pathogenic or pathogenic, and these guidelines are subject to change. In addition, it is our practice to offer support to clinicians and geneticists ordering our tests regarding which genes or panels to order as well as interpretation of genetic variants. We also rely on clinicians to interpret what we report and to incorporate specific information about an individual patient into the physician’s treatment decision.
The marketing, sale and use of our genetic tests could subject us to liability for errors in, misunderstandings of, or inappropriate reliance on, information we provide to clinicians, geneticists or patients, and lead to claims against us if someone were to allege that a test failed to perform as it was designed, if we failed to correctly interpret the test results, if we failed to update the test results due to a reclassification of the variants according to new published guidelines, or if the ordering physician were to misinterpret test results or improperly rely on them when making a clinical decision. In addition, our entry into the reproductive health and pharmacogenetic testing markets expose us to increased liability. A product liability or professional liability claim could result in substantial damages and be costly and time-consuming for us to defend. Although we maintain liability insurance, including for errors and omissions, we cannot assure you that such insurance would fully protect us from the financial impact of defending against these types of claims or any judgments, fines or settlement costs arising out of any such claims. Any liability claim, including an errors and omissions liability claim, brought against us, with or without merit, could increase our insurance rates or prevent us from securing insurance coverage in the future. Additionally, any liability lawsuit could cause injury to our reputation or cause us to suspend sales of our tests. The occurrence of any of these events could have an adverse effect on our reputation and results of operations.
37
Our industry is subject to rapidly changing technology and new and increasing amounts of scientific data related to genes and genetic variants and their role in disease. Our failure to develop tests to keep pace with these changes could make us obsolete.
In recent years, there have been numerous advances in methods used to analyze very large amounts of genomic information and the role of genetics and gene variants in disease and treatment therapies. Our industry has and will continue to be characterized by rapid technological change, increasingly larger amounts of data, frequent new testing service introductions and evolving industry standards, all of which could make our tests obsolete. Our future success will also depend on our ability to keep pace with the evolving needs of our customers on a timely and cost-effective basis and to pursue new market opportunities that develop as a result of technological and scientific advances. Our tests could become obsolete and our business adversely affected unless we continually update our offerings to reflect new scientific knowledge about genes and genetic variations and their role in diseases and treatment therapies.
Our success will depend in part on our ability to generate sales using our internal sales team and through alternative marketing strategies.
We may not be able to market or sell our current tests and any future tests we may develop or acquire effectively enough to drive demand sufficient to support our planned growth. We currently sell our tests primarily through our internal sales force. Historically, our sales efforts have been focused primarily on hereditary cancer and more recently on reproductive health. Our efforts to sell our tests to clinicians and patients outside of oncology may not be successful, or may be difficult to do successfully without significant additional selling and marketing efforts and expense. In addition, following the acquisition of ArcherDX, our sales efforts have expanded to include distributed products sold to laboratories. In the past, we have increased our sales force each year in order to drive our growth, and in October 2020, we increased our sales force through the acquisition of ArcherDX. In addition to the efforts of our sales force, future sales will depend in large part on our ability to develop and substantially expand awareness of our company and our tests through alternative strategies including through education of key opinion leaders, through social media-related and online outreach, education and marketing efforts, and through focused channel partner strategies designed to drive demand for our tests. We also plan to continue to spend on consumer advertising in connection with our direct channel to consumers, which could be costly. We have limited experience implementing these types of marketing efforts. We may not be able to drive sufficient levels of revenue using these sales and marketing methods and strategies necessary to support our planned growth, and our failure to do so could limit our revenue and potential profitability.
We also use a limited number of distributors to assist internationally with sales, logistics, education and customer support. Sales practices utilized by our distributors that are locally acceptable may not comply with sales practices standards required under U.S. laws that apply to us, which could create additional compliance risk. If our sales and marketing efforts are not successful outside the United States, we may not achieve significant market acceptance for our tests outside the United States, which could adversely impact our business.
Impairment in the value of our goodwill or other intangible assets could have a material adverse effect on our operating results and financial condition.
We record goodwill and intangible assets at fair value upon the acquisition of a business. Goodwill represents the excess of amounts paid for acquiring businesses over the fair value of the net assets acquired. Goodwill and indefinite-lived intangible assets are evaluated for impairment annually, or more frequently if conditions warrant, by comparing the carrying value of a reporting unit to its estimated fair value. Intangible assets with definite lives are reviewed for impairment when events or circumstances indicate that their carrying value may not be recoverable. Declines in operating results, divestitures, sustained market declines and other factors that impact the fair value of a reporting unit could result in an impairment of goodwill or intangible assets and, in turn, a charge to net income. Any such charges could have a material adverse effect on our results of operations or financial condition.
38
We rely on a limited number of suppliers or, in some cases, sole suppliers, for some of our laboratory instruments, materials and services, and we may not be able to find replacements or immediately transition to alternative suppliers.
We rely on a limited number of suppliers, or, in some cases, sole suppliers, including Illumina, Inc., Integrated DNA Technologies Incorporated, Qiagen N.V., Roche Holdings Ltd. and Twist Bioscience Corporation for certain laboratory substances used in the chemical reactions incorporated into our processes, which we refer to as reagents, as well as sequencers and other equipment and materials which we use in our laboratory operations. We do not have short- or long-term agreements with most of our suppliers, and our suppliers could cease supplying these materials and equipment at any time, or fail to provide us with sufficient quantities of materials or materials that meet our specifications. Our laboratory operations could be interrupted if we encounter delays or difficulties in securing these reagents, sequencers or other equipment or materials, and if we cannot obtain an acceptable substitute. Any such interruption could significantly affect our business, financial condition, results of operations and reputation. We rely on Illumina as the sole supplier of next generation sequencers and associated reagents and as the sole provider of maintenance and repair services for these sequencers. Any disruption in Illumina’s operations could impact our supply chain and laboratory operations as well as our ability to conduct our tests, and it could take a substantial amount of time to integrate replacement equipment into our laboratory operations.
We believe that there are only a few other manufacturers that are currently capable of supplying and servicing the equipment necessary for our laboratory operations, including sequencers and various associated reagents. The use of equipment or materials provided by these replacement suppliers would require us to alter our laboratory operations. Transitioning to a new supplier would be time consuming and expensive, may result in interruptions in our laboratory operations, could affect the performance specifications of our laboratory operations or could require that we revalidate our tests. We cannot assure you that we will be able to secure alternative equipment, reagents and other materials, and bring such equipment, reagents and materials on line and revalidate them without experiencing interruptions in our workflow. In the case of an alternative supplier for Illumina, we cannot assure you that replacement sequencers and associated reagents will be available or will meet our quality control and performance requirements for our laboratory operations. If we encounter delays or difficulties in securing, reconfiguring or revalidating the equipment and reagents we require for our tests, our business, financial condition, results of operations and reputation could be adversely affected.
Our planned STRATAFIDE and PCM products are currently being developed to use only Illumina’s sequencing platform. Without access to these sequencers, we would be unable commercialize these products. In addition, any efforts to validate these distributed products on additional sequencing platforms would require significant resources, expenditures and time and attention of our management, and there is no guarantee that we will be successful in implementing any such sequencing platforms in a commercially sustainable way. We also cannot guarantee that it will appropriately prioritize or select alternative sequencing platforms on which to focus our efforts.
If our laboratories become inoperable due to disasters, health epidemics or for any other reasons, we will be unable to perform our tests and our business will be harmed.
We perform all of our tests at our production facilities in San Francisco and Irvine, California, in Golden, Colorado and in Seattle, Washington. Our laboratories and the equipment we use to perform our tests would be costly to replace and could require substantial lead time to replace and qualify for use. Our laboratories may be harmed or rendered inoperable by natural or man-made disasters, including earthquakes, flooding, fire and power outages, or by health epidemics, such as the COVID-19 pandemic, which may render it difficult or impossible for us to perform our tests for some period of time. This risk of natural disaster is especially high for us since we perform the substantial majority of our tests at our San Francisco laboratory, which is located in an active seismic region, and we do not have a redundant facility to perform the same tests in the event our San Francisco laboratory is inoperable. The inability to perform our tests or the backlog that could develop if our laboratories are inoperable for even a short period of time may result in the loss of customers or harm our reputation. Although we maintain insurance for damage to our property and the disruption of our business, this insurance may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, if at all.
39
ArcherDX relies on third-party laboratories to perform portions of its service offerings.
A large portion of ArcherDX’s biopharmaceutical testing services is performed by third-party laboratories while the remaining portion is performed by third-party laboratories certified under the CLIA, or our CLIA-certified laboratory in Colorado. The third-party laboratories are subject to contractual obligations to perform these services for us, but are not otherwise under our control. We therefore do not control the capacity and quality control efforts of these third-party laboratories other than through our ability to enforce contractual obligations on volume and quality systems, and have no control over such laboratories’ compliance with applicable legal and regulatory requirements. We also have no control over the timeliness of such laboratories’ performance of their obligations to us, and the third-party laboratories that we have contracted with have in the past had, and occasionally continue to have, issues with delivering results to us or resolving issues with us within the time frames we expected or established in our contracts with them, which sometimes results in longer than expected turnaround times for, or negatively impacts the performance of, these tests and services. In the event of any adverse developments with these third-party laboratories or their ability to perform their obligations in a timely manner and in accordance with the standards that we and our customers expect, our ability to service customers may be delayed, interrupted or otherwise adversely affected, which could result in a loss of customers and harm to our reputation. Furthermore, when these issues arise, we have had to expend time, management’s attention and other resources to address and remedy such issues.
We may not have sufficient alternative backup if one or more of the third-party laboratories that we contract with are unable to satisfy their obligations to us with sufficient performance, quality and timeliness. Any natural or other disaster, acts of war or terrorism, shipping embargoes, labor unrest or political instability or similar events at one or more of these third-party laboratories’ facilities that causes a loss of capacity would heighten the risks that we face. Changes to or termination of agreements or inability to renew agreements with these third-party laboratories or enter into new agreements with other laboratories that are able to perform such portions of our service offerings could impair, delay or suspend our efforts to market and sell these services.
The loss of any member or change in structure of our senior management team could adversely affect our business.
Our success depends in large part upon the skills, experience and performance of members of our executive management team and others in key leadership positions. The efforts of these persons will be critical to us as we continue to develop our technologies and test processes and focus on scaling our business. If we were to lose one or more key executives, we may experience difficulties in competing effectively, developing our technologies and implementing our business strategy. All of our executives and employees are at-will, which means that either we or the executive or employee may terminate their employment at any time. We do not carry key person insurance for any of our executives or employees. In addition, we do not have a long-term retention agreement in place with our president and chief executive officer.
Development of new tests is a complex process, and we may be unable to commercialize new tests on a timely basis, or at all.
We cannot assure you that we will be able to develop and commercialize new tests on a timely basis. Before we can commercialize any new tests, we will need to expend significant funds in order to:
•conduct research and development;
•further develop and scale our laboratory processes; and
•further develop and scale our infrastructure to be able to analyze increasingly larger and more diverse amounts of data.
Our testing service development process involves risk, and development efforts may fail for many reasons, including:
•failure of any test to perform as expected;
•lack of validation or reference data; or
•failure to demonstrate utility of a test.
As we develop tests, we will have to make significant investments in development, marketing and selling resources. In addition, competitors may develop and commercialize competing tests faster than we are able to do so.
40
Our research and development efforts to add additional indications to our IVD products, if approved, will be hindered if we are not able to contract with third parties for access to tissue samples.
Under standard clinical practice, tumor biopsies removed from patients are preserved and stored in formalin-fixed paraffin embedded, or FFPE, format, and liquid biopsies are taken with a blood draw and stored in blood collection tubes. In order to add additional indications to our IVD products, if approved, we will need to secure access to these FFPE tumor biopsy and liquid biopsy samples, as well as information pertaining to the clinical outcomes of the patients from which they were derived for its IVD development activities. Others compete with us for access to these samples. Additionally, the process of negotiating access to samples is lengthy because it typically involves numerous parties and approval levels to resolve complex issues such as usage rights, institutional review board approval, privacy rights, publication rights, intellectual property ownership and research parameters. If we are unable to negotiate access to tissue samples on a timely basis or on commercially reasonable terms, or at all, or if other laboratories or competitors secure access to these samples before us, our ability to research, develop and commercialize future IVD products will be limited or delayed.
We depend on our information technology systems, and any failure of these systems could harm our business.
We depend on information technology and telecommunications systems for significant elements of our operations, including our laboratory information management system, our bioinformatics analytical software systems, our database of information relating to genetic variations and their role in disease process and drug metabolism, our clinical report optimization systems, our customer-facing web-based software, our customer reporting, and our various customer tools and platforms. We have installed, and expect to expand, a number of enterprise software systems that affect a broad range of business processes and functional areas, including for example, systems handling human resources, financial controls and reporting, customer relationship management, regulatory compliance and other infrastructure operations. In addition, we intend to extend the capabilities of both our preventative and detective security controls by augmenting the monitoring and alerting functions, the network design, and the automatic countermeasure operations of our technical systems. These information technology and telecommunications systems support a variety of functions, including laboratory operations, test validation, sample tracking, quality control, customer service support, billing and reimbursement, research and development activities, scientific and medical curation, and general administrative activities, including financial reporting.
Information technology and telecommunications systems are vulnerable to damage from a variety of sources, including telecommunications or network failures, malicious human acts and natural disasters. Moreover, despite network security and back-up measures, some of our servers are potentially vulnerable to physical or electronic break-ins, computer viruses and similar disruptive problems. Despite the precautionary measures we have taken to prevent unanticipated problems that could affect our information technology and telecommunications systems, failures or significant downtime of our information technology or telecommunications systems or those used by our third-party service providers could prevent us from conducting tests, preparing and providing reports to clinicians, billing payers, processing reimbursement appeals, handling physician or patient inquiries, conducting research and development activities, and managing the administrative and financial aspects of our business. Any disruption or loss of information technology or telecommunications systems on which critical aspects of our operations depend could have an adverse effect on our business and results of operations.
Technical problems have arisen, and may arise in the future, in connection with our data and systems, including those that are hosted by third-party providers, which have in the past and may in the future result in interruptions in our business and operations. These types of problems may be caused by a variety of factors, including infrastructure changes, human or software errors, viruses, security attacks, fraud, spikes in customer usage and denial of service issues. From time to time, large third-party web hosting providers have experienced outages or other problems that have resulted in their systems being offline and inaccessible. Such outages could materially impact our business and operations.
41
Ethical, legal and social concerns related to the use of genetic information could reduce demand for our tests.
Genetic testing has raised ethical, legal and social issues regarding privacy rights and the appropriate uses of the resulting information. Governmental authorities could, for social or other purposes, limit or regulate the use of genetic information or genetic testing or prohibit testing for genetic predisposition to certain conditions, particularly for those that have no known cure. Similarly, these concerns may lead patients to refuse to use, or clinicians to be reluctant to order, genomic tests even if permissible. These and other ethical, legal and social concerns may limit market acceptance of our tests or reduce the potential markets for our tests, either of which could have an adverse effect on our business, financial condition or results of operations.
Our international business exposes us to business, regulatory, political, operational, financial and economic risks associated with doing business outside of the United States.
Doing business internationally involves a number of risks, including:
•multiple, conflicting and changing laws and regulations such as privacy regulations, tax laws, export and import restrictions, employment laws, regulatory requirements, and other governmental approvals, permits and licenses;
•failure by us or our distributors to obtain regulatory approvals for the use of our tests in various countries;
•complexities and difficulties in obtaining protection and enforcing our intellectual property;
•difficulties in staffing and managing foreign operations;
•complexities associated with managing multiple payer reimbursement regimes, government payers or patient self-pay systems;
•logistics and regulations associated with shipping samples, including infrastructure conditions, customs and transportation delays;
•limits on our ability to penetrate international markets if we do not to conduct our tests locally;
•natural disasters, including the recent and ongoing outbreak and spreading of Coronavirus, political and economic instability, including wars, terrorism and political unrest, outbreak of disease, boycotts, curtailment of trade and other business restrictions; and
•regulatory and compliance risks that relate to maintaining accurate information and control over activities that may fall within the purview of the U.S. Foreign Corrupt Practices Act, or FCPA, its books and records provisions, or its anti-bribery provisions.
Any of these factors could significantly harm our international operations and, consequently, our revenue and results of operations.
In addition, applicable export or import laws and regulations such as prohibitions on the export of samples imposed by countries outside of the United States, or international privacy or data restrictions that are different or more stringent than those of the United States, may require that we build additional laboratories or engage in joint ventures or other business partnerships in order to offer our tests internationally in the future. Any such restrictions would impair our ability to offer our tests in such countries and could have an adverse effect on our business, financial condition and results of operations.
42
Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.
At December 31, 2020, our total gross deferred tax assets were $418.9 million. Due to our lack of earnings history and uncertainties surrounding our ability to generate future taxable income, our net deferred tax assets have been fully offset by a valuation allowance. The deferred tax assets are primarily comprised of federal and state tax net operating losses and tax credit carryforwards. Furthermore, under Section 382 of the Internal Revenue Code of 1986, as amended, or the Internal Revenue Code, if a corporation undergoes an “ownership change,” the corporation’s ability to use its pre-change net operating loss carryforwards, or NOLs, and other pre-change tax attributes (such as research tax credits) to offset its future taxable income may be limited. In general, an “ownership change” occurs if there is a cumulative change in our ownership by “5% shareholders” that exceeds 50 percentage points over a rolling three-year period. Some of our prior acquisitions have resulted in an ownership change, and we may experience ownership changes in the future. Our existing NOLs and tax credit carryovers may be subject to limitations arising from previous ownership changes, and if we undergo one or more ownership changes in connection with completed acquisitions, or other future transactions in our stock, our ability to utilize NOLs and tax credit carryovers could be further limited by Section 382 of the Internal Revenue Code. As a result, if we earn net taxable income, our ability to use our pre-change net operating loss and tax credit carryforwards to offset U.S. federal taxable income may be subject to limitations, which could potentially result in increased future tax liability to us. The annual limitation may result in the expiration of certain net operating loss and tax credit carryforwards before their utilization. In addition, the Tax Cuts and Jobs Act limits the deduction for NOLs to 80% of current year taxable income and eliminates NOL carrybacks. Also, at the state level, there may be periods during which the use of NOLs is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed.
Risks related to government regulation
If the FDA regulates the tests we currently offer as LDTs as medical devices, we could incur substantial costs and our business, financial condition and results of operations could be adversely affected.
We provide many of our tests as laboratory-developed tests, or LDTs. CMS and certain state agencies regulate the performance of LDTs (as authorized by CLIA, and state law, respectively).
Historically, the FDA has exercised enforcement discretion with respect to most LDTs and has not required laboratories that furnish LDTs to comply with the agency’s requirements for medical devices (e.g., establishment registration, device listing, quality systems regulations, premarket clearance or premarket approval, and post-market controls). In recent years, however, the FDA has stated it intends to end its policy of general enforcement discretion and regulate certain LDTs as medical devices. To this end, on October 3, 2014, the FDA issued two draft guidance documents, entitled “Framework for Regulatory Oversight of Laboratory Developed Tests (LDTs)” and “FDA Notification and Medical Device Reporting for Laboratory Developed Tests (LDTs),” respectively, that set forth a proposed risk-based regulatory framework that would apply varying levels of FDA oversight to LDTs. Subsequently, on January 13, 2017, the FDA published a “discussion paper” in which it outlined a substantially revised “possible approach” to the oversight of LDTs.
In March 2020, a bill titled the “Verifying Accurate Leading-edge IVCT Development Act of 2020,” or VALID Act, was officially introduced in Congress. The bill proposes a risk-based approach to regulate LDTs and creates a new in vitro clinical test, or IVCT, category of regulated products, which includes LDTs, and a regulatory structure under the FDA. As proposed, the bill grandfathers many existing tests from the proposed premarket approval, quality systems, and labeling requirements, respectively, but would require such tests to comply with other regulatory requirements (e.g., registration and listing, adverse event reporting). Later that month, Senator Paul introduced the Verified Innovative Testing in American Laboratories Act of 2020, or VITAL Act, which proposes that all aspects of “laboratory-developed testing procedures” be subject to regulation under CLIA, and that no aspects of such procedures be subject to regulation by the FDA. We cannot predict if either of these bills will be enacted in their current (or any other) form and cannot quantify the effect of these bills on our business.
In August 2020, the U.S. Department of Health and Human Services, the parent agency for FDA, announced that the FDA “will not require premarket review of LDTs absent notice-and-comment rulemaking, as opposed to through guidance documents, compliance manuals, website statements, or other informal issuances.” It is unclear at this time whether this policy will be retained by the Biden Administration, and if so, when the FDA might seek to begin the notice and comment rulemaking process.
Legislative proposals addressing the FDA’s oversight of LDTs have been introduced in previous Congresses, and we expect that new legislative proposals may be introduced from time-to-time. The likelihood that Congress will pass such legislation and the extent to which such legislation may affect the FDA’s plans to regulate certain LDTs as medical devices is difficult to predict at this time.
43
In April 2020, we completed our acquisition of Genelex Solutions, LLC, which offers certain pharmacogenetic, or PGx, tests as LDTs. Recently the FDA has taken a more active role in the oversight of PGx tests offered as LDTs. In 2019, the FDA contacted several clinical laboratories, including Genelex, to demand changes to PGx test reports and marketing materials. In February 2020, the FDA issued a statement indicating that it continues to have concerns about the claims that certain clinical laboratories make with respect to their PGx tests, and published tables that list PGx associations for which FDA has determined that the data support therapeutic management recommendations, a potential impact on safety or response, or a potential impact on pharmacokinetic properties only, respectively. To date, however, the FDA has not provided any general guidance on the type(s) of claims or other characteristics that will cause a PGx test to fall outside FDA’s enforcement discretion. As such, the extent to which the FDA will allow any laboratory, including Genelex or Invitae, to offer PGx tests in their current form without meeting FDA regulatory requirements for medical devices is unclear at this time.
If the FDA ultimately regulates certain LDTs (either as medical devices or as part of a new stand-alone regulatory category for IVCTs), whether via individualized enforcement action, or more generally, as outlined in final guidance or final regulation, or as instructed by Congress, our tests may be subject to certain additional regulatory requirements. Complying with the FDA’s requirements can be expensive, time-consuming and subject us to significant or unanticipated delays. Insofar as we may be required to obtain premarket clearance or approval to perform or continue performing an LDT, we cannot assure you that we will be able to obtain such authorization. Even if we obtain regulatory clearance or approval where required, such authorization may not be for the intended uses that we believe are commercially attractive or are critical to the commercial success of our tests. As a result, the application of the FDA’s requirements to our tests could materially and adversely affect our business, financial condition and results of operations.
Failure to comply with applicable FDA regulatory requirements may trigger a range of enforcement actions by the FDA including warning letters, civil monetary penalties, injunctions, criminal prosecution, recall or seizure, operating restrictions, partial suspension or total shutdown of operations, and denial of or challenges to applications for clearance or approval, as well as significant adverse publicity.
In addition, in November 2013, the FDA issued final guidance regarding the distribution of products labeled for research use only. Certain of the reagents and other products we use in our tests are labeled as research use only products. Certain of our suppliers may cease selling research use only products to us and any failure to obtain an acceptable substitute could significantly and adversely affect our business, financial condition and results of operations.
If we fail to comply with federal, state and foreign laboratory licensing requirements, we could lose the ability to perform our tests or experience disruptions to our business.
We are subject to CLIA, a federal law that regulates clinical laboratories that perform testing on specimens derived from humans for the purpose of providing information for the diagnosis, prevention or treatment of disease. CLIA regulations establish specific standards with respect to personnel qualifications, facility administration, proficiency testing, quality control, quality assurance and inspections. CLIA certification is also required in order for us to be eligible to bill state and federal healthcare programs, as well as many private third-party payers, for our tests. We have current CLIA certifications to conduct our tests at our laboratories in San Francisco and Irvine, California, Golden, Colorado, and Seattle, Washington. To renew these certifications, we are subject to survey and inspection every two years. Moreover, CLIA inspectors may make random inspections of our clinical reference laboratories.
We are also required to maintain in-state licenses to conduct testing in California and Washington. California and Washington laws establish standards for day-to-day operation of our clinical reference laboratories in San Francisco and Irvine, and in Seattle, respectively, which include the training and skills required of personnel and quality control. (Our Colorado laboratory is not required to maintain a state clinical laboratory license.)
Several states require the licensure of out‑of‑state laboratories that accept specimens from those states. Our California laboratories hold the required out‑of‑state laboratory licenses for Maryland, New York, Pennsylvania, and Rhode Island. Our Washington laboratory holds the required out-of-state laboratory licenses in Maryland, New York, Pennsylvania, and Rhode Island (but not California). Our laboratory in Colorado holds the required out‑of‑state laboratory licenses for California, Maryland, Pennsylvania and Rhode Island (but not New York).
44
In addition to having laboratory licenses in New York, our clinical reference laboratories are approved on test-specific bases for the tests they run as LDTs by the New York State Department of Health, or NYDOH, for tests offered to patients in New York. Other states may adopt similar licensure requirements in the future, which may require us to modify, delay or stop our operations in such jurisdictions. We may also be subject to regulation in foreign jurisdictions as we seek to expand international utilization of our tests or such jurisdictions adopt new licensure requirements, which may require review of our tests in order to offer them or may have other limitations such as restrictions on the transport of samples necessary for us to perform our tests that may limit our ability to make our tests available outside of the United States. Complying with licensure requirements in new jurisdictions may be expensive, time-consuming, and subject us to significant and unanticipated delays.
In order to eventually market certain of our current or future products and services in any particular foreign jurisdiction, we must establish and comply with numerous and varying regulatory requirements on a jurisdiction-by-jurisdiction basis regarding quality, safety, performance and efficacy. In addition, clinical trials or clinical investigations conducted in one country may not be accepted by regulatory authorities in other countries, and regulatory clearance, authorization or approval in one country does not guarantee regulatory clearance, authorization or approval in any other country. For example, the performance characteristics of certain of our products and services may need to be validated separately in specific ethnic and genetic populations. Approval processes vary among countries and can involve additional product testing and validation and additional administrative review periods.
Seeking foreign regulatory clearance, authorization or approval could result in difficulties and costs for us and our collaborators and require additional preclinical studies, clinical trials or clinical investigations which could be costly and time-consuming. Regulatory requirements and ethical approval obligations can vary widely from country to country and could delay or prevent the introduction of certain of our products and services in those countries. The foreign regulatory clearance, authorization or approval process involves all of the risks and uncertainties associated with FDA clearance, authorization or approval. ArcherDX currently sells its RUO products outside the United States but has no experience in obtaining regulatory clearance, authorization or approval in international markets other than Japan. If we or our collaborators fail to comply with regulatory requirements in international markets or to obtain and maintain required regulatory clearances, authorizations or approvals in international markets, or if those approvals are delayed, our target market will be reduced and our ability to realize the full market potential of our products and services will be unrealized.
Failure to comply with applicable clinical laboratory licensure requirements may result in a range of enforcement actions, including license suspension, limitation, or revocation, directed plan of action, onsite monitoring, civil monetary penalties, criminal sanctions, and cancellation of the laboratory’s approval to receive Medicare and Medicaid payment for its services, as well as significant adverse publicity. Any sanction imposed under CLIA, its implementing regulations, or state or foreign laws or regulations governing clinical laboratory licensure, or our failure to renew our CLIA certifications, a state or foreign license, or accreditation, could have a material adverse effect on our business, financial condition and results of operations. Even if we were able to bring our laboratory back into compliance, we could incur significant expenses and potentially lose revenue in doing so.
The College of American Pathologists, or CAP, maintains a clinical laboratory accreditation program. CAP asserts that its program is “designed to go well beyond regulatory compliance” and helps laboratories achieve the highest standards of excellence to positively impact patient care. While not required to operate a CLIA-certified laboratory, many private insurers require CAP accreditation as a condition to contracting with clinical laboratories to cover their tests. In addition, some countries outside the United States require CAP accreditation as a condition to permitting clinical laboratories to test samples taken from their citizens. We have CAP accreditations for our laboratories. Failure to maintain CAP accreditation could have a material adverse effect on the sales of our tests and the results of our operations.
45
We may not be able to obtain regulatory clearance or approval of our IVD products, or even if approved, such products may not be approved for guideline inclusion, which could adversely our ability to realize the intended benefits of our recently completed acquisition of ArcherDX.
A significant portion of ArcherDX’s commercial strategy, including for STRATAFIDE and PCM, relies on receiving regulatory approvals with guideline inclusion to strengthen its position in establishing coverage and reimbursement of its IVD products with both public and private payers. If we do not receive such regulatory approvals in a timely manner or at all, or we are not successful in obtaining such guideline inclusion, it may not be able to commercialize our IVD products. Additionally, third-party payers may be unwilling to provide sufficient coverage and reimbursement for these products necessary for hospitals and other healthcare providers to adopt our solutions as part of their oncological treatment strategy. ArcherDX has also focused its efforts on the development of PCM for FDA clearance and approval as a prognostic device for predicting recurrence of a primary cancer after initial treatment, which can include surgery alone or surgery plus adjuvant therapy.
Moreover, development of the data necessary to obtain regulatory clearance and/or approval of an IVD, such as STRATAFIDE, is time-consuming and carries with it the risk of not yielding the desired results. The performance achieved in published studies may not be repeated in later studies that may be required to obtain FDA clearance and/or approval or regulatory approvals in foreign jurisdictions. Limited results from earlier-stage verification studies may not predict results from studies in larger numbers of subjects drawn from more diverse populations over longer periods of time. Unfavorable results from ongoing preclinical and clinical studies could result in delays, modifications or abandonment of ongoing analytical or future clinical studies, or abandonment of a product development program, or may delay, limit or prevent regulatory approvals or clearances or commercialization of our product candidates, any of which may materially impact our ability to realize the expected benefits of our recently completed acquisition of ArcherDX.
Complying with numerous statutes and regulations pertaining to our business is an expensive and time-consuming process, and any failure to comply could result in substantial penalties.
Our operations are subject to other extensive federal, state, local and foreign laws and regulations, all of which are subject to change. These laws and regulations currently include, among others:
•HIPAA, which establishes comprehensive federal standards with respect to the privacy and security of protected health information and requirements for the use of certain standardized electronic transactions;
•amendments to HIPAA under HITECH, which strengthen and expand HIPAA privacy and security compliance requirements, increase penalties for violators and expand vicarious liability, extend enforcement authority to state attorneys general, and impose requirements for breach notification;
•the GDPR, which imposes strict privacy and security requirements on controllers and processors of personal data, including enhanced protections for “special categories” of personal data, including sensitive information such as health and genetic information of data subjects;
•the CCPA, which, among other things, regulates how subject businesses may collect, use, and disclose the personal information of consumers who reside in California, affords rights to consumers that they may exercise against businesses that collect their information, and requires implementation of reasonable security measures to safeguard personal information of California consumers;
•the federal Anti-Kickback Statute, which prohibits knowingly and willfully offering, paying, soliciting or receiving remuneration, directly or indirectly, overtly or covertly, in cash or in kind, to induce or in return for the referral of an individual, for the furnishing of or arrangement for the furnishing of any item or service for which payment may be made in whole or in part by a federal healthcare program, or the purchasing, leasing, ordering, arranging for, or recommend purchasing, leasing or ordering, any good, facility, item or service for which payment may be made, in whole or in part, under a federal healthcare program;
•EKRA, which prohibits payments for referrals to recovery homes, clinical treatment facilities, and laboratories and reaches beyond federal health care programs, to include private insurance;
•the federal physician self-referral law, known as the Stark Law, which prohibits a physician from making a referral to an entity for certain designated health services covered by the Medicare program, including laboratory and pathology services, if the physician or an immediate family member has a financial relationship with the entity unless an exception applies, and prohibits an entity from billing for designated health services furnished pursuant to a prohibited referral;
46
•the federal false claims law, which imposes liability on any person or entity that, among other things, knowingly presents, or causes to be presented, a false or fraudulent claim for payment to the federal government;
•the federal Civil Monetary Penalties Law, which prohibits, among other things, the offering or transfer of remuneration to a Medicare or state healthcare program beneficiary if the person knows or should know it is likely to influence the beneficiary’s selection of a particular provider, practitioner or supplier of services reimbursable by Medicare or a state healthcare program, unless an exception applies;
•the HIPAA fraud and abuse provisions, which create new federal criminal statutes that prohibit, among other things, defrauding health care benefit programs, willfully obstructing a criminal investigation of a healthcare offense and falsifying or concealing a material fact or making any materially false statements in connection with the payment for healthcare benefits, items or services;
•other federal and state fraud and abuse laws, such as anti-kickback laws, prohibitions on self-referral, fee-splitting restrictions, insurance fraud laws, anti-markup laws, prohibitions on the provision of tests at no or discounted cost to induce physician or patient adoption, and false claims acts, which may extend to services reimbursable by any third-party payer, including private insurers;
•the 21st Century Cures Act information blocking prohibition, which prohibits covered actors from engaging in certain practices that are likely to interfere with the access, exchange, or use of electronic health information;
•the Physician Payments Sunshine Act and similar state laws that require reporting of certain payments and other transfers of value made by applicable manufacturers, directly or indirectly, to or on behalf of covered recipients including physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals as well as ownership and investment interests held by physicians and their immediate family members. Beginning in 2022, applicable manufacturers also will be required to report such information regarding its relationships with physician assistants, nurse practitioners, clinical nurse specialists, certified registered nurse anesthetists and certified nurse midwives during the previous year;
•state laws that limit or prohibit the provision of certain payments and other transfers of value to certain covered healthcare providers;
•the prohibition on reassignment of Medicare claims, which, subject to certain exceptions, precludes the reassignment of Medicare claims to any other party;
•state laws that prohibit other specified practices, such as billing clinicians for testing that they order; waiving coinsurance, copayments, deductibles and other amounts owed by patients; billing a state Medicaid program at a price that is higher than what is charged to one or more other payers; and
•similar foreign laws and regulations that apply to us in the countries in which we operate or may operate in the future.
We have adopted policies and procedures designed to comply with these laws and regulations. In the ordinary course of our business, we conduct internal reviews of our compliance with these laws. Our compliance may also be subject to governmental review. The growth of our business and our expansion outside of the United States may increase the potential of violating these laws or our internal policies and procedures. The risk of our being found in violation of these or other laws and regulations is further increased by the fact that many have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations. Any action brought against us for violation of these or other laws or regulations, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. If our operations are found to be in violation of any of these laws and regulations, we may be subject to any applicable penalty associated with the violation, including significant administrative, civil and criminal penalties, damages, fines, imprisonment, exclusion from participation in Federal healthcare programs, refunding of payments received by us, and curtailment or cessation of our operations. Any of the foregoing consequences could seriously harm our business and our financial results.
47
Healthcare policy changes, including legislation reforming the U.S. healthcare system, may have a material adverse effect on our financial condition, results of operations and cash flows.
In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, collectively referred to as the Affordable Care Act, was enacted in the United States, which made a number of substantial changes in the way healthcare is financed by both governmental and private insurers. Policy changes or implementation of new health care legislation could result in significant changes to health care systems. In the United States, this could include potential modification or repeal of all or parts of the Affordable Care Act.
In April 2014, Congress passed the Protecting Access to Medicare Act of 2014, or PAMA, which included substantial changes to the way in which clinical laboratory services are paid under Medicare. Under PAMA (as amended by the Further Consolidated Appropriations Act, 2020 and the Coronavirus Aid, Relief, and Economic Security Act, respectively) and its implementing regulations, clinical laboratories must report to CMS private payer rates beginning in 2017, and then in 2022 and every three years thereafter for clinical diagnostic laboratory tests that are not advanced diagnostic laboratory tests and every year for advanced diagnostic laboratory tests.
We do not believe that our tests meet the definition of advanced diagnostic laboratory tests, but in the event that we seek designation for one or more of our tests as an advanced diagnostic laboratory test and the tests are determined by CMS to meet these criteria or new criteria developed by CMS, we would be required to report private payer data for those tests annually. Otherwise, we will be required to report private payer rates for our tests on an every three years basis starting in 2022. Laboratories that fail to timely report the required payment information may be subject to substantial civil money penalties.
As set forth in the PAMA final rule, for tests furnished on or after January 1, 2018, Medicare payments for clinical diagnostic laboratory tests are paid based upon these reported private payer rates. For clinical diagnostic laboratory tests that are assigned a new or substantially revised code, initial payment rates for clinical diagnostic laboratory tests that are not advanced diagnostic laboratory tests will be assigned by the cross-walk or gap-fill methodology. Initial payment rates for new advanced diagnostic laboratory tests will be based on the actual list charge for the laboratory test. The payment rates calculated under PAMA went into effect starting January 1, 2018. Where applicable, reductions to payment rates resulting from the new methodology are limited to 10% per test per year in each of the years 2018 through 2020. Rates will be held at 2020 levels during 2021, and then, where applicable based upon median private payer rates reported in 2017 or 2022, reduced by up to 15% per test per year in each of 2022 through 2024 (with a second round of private payer rate reporting in 2022 to establish rates for 2023 through 2025).
PAMA also authorized the adoption of new, temporary billing codes and/or unique test identifiers for FDA-cleared or approved tests as well as advanced diagnostic laboratory tests. The CPT® Editorial Panel approved a proposal to create a new section of billing codes to facilitate implementation of this section of PAMA, but these codes would apply to our tests only if we apply for such codes.
In March 2018, CMS published a national coverage determination, or NCD, for next generation sequencing, or NGS tests for somatic (acquired) cancer testing. CMS subsequently updated this NCD in January 2020 to address coverage for NGS tests for germline (inherited) cancer testing and to clarify certain aspects of Medicare’s coverage of NGS for somatic cancer testing. For somatic cancer testing, the updated NCD establishes full coverage for FDA-approved or FDA-cleared NGS-based companion diagnostic assays that report results using report templates that specify treatment options when offered for their FDA-approved or FDA-cleared use(s), ordered by the patient’s treating physician for Medicare beneficiaries with advanced cancer (recurrent, relapsed, refractory, metastatic, or advanced stage III or IV cancer) who have not have previously been tested with the same test using NGS for the same cancer genetic content, and have decided to seek further cancer treatment (e.g., therapeutic chemotherapy). The NCD also gives MACs the authority to establish local coverage for NGS-based somatic cancer assays that are not FDA-approved or FDA-cleared companion diagnostics when offered to patients meeting the above-referenced criteria. It appears that NGS-based somatic cancer tests provided for patients with cancer that do not meet the above-referenced criteria, e.g., patients with earlier stage cancers, are currently nationally non-covered under the NCD.
48
Effective January 27, 2020, the NCD also established full coverage for FDA-approved or FDA-cleared NGS-based germline tests that report results using report templates that specify treatment options when ordered by the patient’s treating physician for patients with ovarian or breast cancer, a clinical indication for germline testing for hereditary breast or ovarian cancer, and a risk factor for germline breast or ovarian cancer, provided the patient has not previously been tested with the same germline test using NGS for the same germline genetic content. The NCD also gives MACs the authority to establish local coverage for NGS-based germline tests for ovarian or breast cancer that are not FDA-approved or FDA-cleared, as well as for NGS-based tests for any other cancer diagnosis (regardless of the test’s FDA regulatory status) when offered to patients meeting the above-referenced criteria for germline testing. Since we already have local coverage for our germline tests for ovarian and breast cancer, we believe that the NCD will not have a material impact on which of our tests will be reimbursable by CMS for Medicare patients.
We cannot predict whether future healthcare initiatives will be implemented at the federal or state level, or how any future legislation or regulation may affect us. For instance, the payment reductions imposed by the Affordable Care Act and the expansion of the federal and state governments’ role in the U.S. healthcare industry as well as changes to the reimbursement amounts paid by payers for our tests and future tests or our medical procedure volumes may reduce our profits and have a materially adverse effect on our business, financial condition, results of operations and cash flows. Notably, Congress enacted legislation in 2017 that eliminated the Affordable Care Act’s “individual mandate” beginning in 2019, which may significantly impact the number of covered lives participating in exchange plans. The U.S. Supreme Court is currently reviewing the constitutionality of the Affordable Care Act, although it is unclear when a decision will be made. Moreover, Congress has proposed on several occasions to impose a 20% coinsurance on patients for clinical laboratory tests reimbursed under the clinical laboratory fee schedule, which would increase our billing and collecting costs and decrease our revenue. Further, it is possible that additional governmental action be taken in response to the COVID-19 pandemic.
If we use hazardous materials in a manner that causes injury, we could be liable for resulting damages.
Our activities currently require the use of hazardous chemicals and biological material. We cannot eliminate the risk of accidental contamination or injury to employees or third parties from the use, storage, handling or disposal of these materials. In the event of contamination or injury, we could be held liable for any resulting damages, and any liability could exceed our resources or any applicable insurance coverage we may have. In 2018, we decommissioned our laboratory in Massachusetts; however, we could be held liable for any damages resulting from our prior use of hazardous chemicals and biological materials at this facility. Additionally, we are subject on an ongoing basis to federal, state and local laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. The cost of compliance with these laws and regulations may become significant, and our failure to comply may result in substantial fines or other consequences, and either could negatively affect our operating results.
We could be adversely affected by violations of the FCPA and other worldwide anti-bribery laws.
We are subject to the FCPA, which prohibits companies and their intermediaries from making payments in violation of law to non-U.S. government officials for the purpose of obtaining or retaining business or securing any other improper advantage. We are increasing our direct sales and operations personnel outside the United States, in which we have limited experience. We use a limited number of independent distributors to sell our tests internationally, which requires a high degree of vigilance in maintaining our policy against participation in corrupt activity, because these distributors could be deemed to be our agents, and we could be held responsible for their actions. Other U.S. companies in the medical device and pharmaceutical fields have faced criminal penalties under the FCPA for allowing their agents to deviate from appropriate practices in doing business with these individuals. We are also subject to similar anti-bribery laws in the jurisdictions in which we operate, including the United Kingdom’s Bribery Act of 2010, which also prohibits commercial bribery and makes it a crime for companies to fail to prevent bribery. These laws are complex and far-reaching in nature, and, as a result, we cannot assure you that we would not be required in the future to alter one or more of our practices to be in compliance with these laws or any changes in these laws or the interpretation thereof. Any violations of these laws, or allegations of such violations, could disrupt our operations, involve significant management distraction, involve significant costs and expenses, including legal fees, and could result in a material adverse effect on our business, prospects, financial condition or results of operations. We could also incur severe penalties, including criminal and civil penalties, disgorgement and other remedial measures.
49
Risks related to our intellectual property
One of ArcherDX’s competitors has alleged that its Anchored Multiplex PCR, or AMP, chemistry and products using AMP are infringing on its intellectual property, and ArcherDX may be required to redesign the technology, obtain a license, cease using the AMP chemistry altogether and/or pay significant damages, among other consequences, any of which would have a material adverse effect on ArcherDX’s business as well as our financial condition and results of operations, and the intended benefits of our recently completed acquisition of ArcherDX.
ArcherDX’s AMP chemistry underlies all of its RUO products and is also the foundation of STRATAFIDE and Personalized Cancer Monitoring, or PCM. On January 27, 2020, one of ArcherDX’s competitors, Natera, Inc., or Natera, filed a complaint against ArcherDX in the United States District Court for the District of Delaware, alleging that ArcherDX’s products using AMP chemistry, and the manufacture, use, sale, and offer for sale of such products, infringe U.S. Patent No. 10,538,814. On April 15, 2020, Natera amended its complaint to allege that ArcherDX’s products using AMP chemistry and the manufacture, use, sale, and offer for sale of such products, infringe U.S. Patent No. 10,538,814, U.S. Patent No. 10,557,172, U.S. Patent No. 10,590,482, and U.S. Patent No. 10,597,708, each of which are held by Natera. On August 6, 2020, Natera filed another complaint against ArcherDX in the United States District Court for the District of Delaware alleging that ArcherDX’s products using AMP chemistry, and the manufacture, use, sale, and offer for sale of such products, infringe U.S. Patent No. 10,731,220 (together with U.S. Patent Nos. 10,538,814, 10,557,172, 10,590,482, and 10,597,708, the “Natera Asserted Patents.”) Natera seeks, among other things, damages and other monetary relief, costs and attorneys’ fees, and an order enjoining ArcherDX from further infringement of The Natera Asserted Patents. On October 13, 2020, the court issued an order denying ArcherDX's motion for dismissal of Natera’s infringement claims against STRATAFIDE, PCM, and ArcherMET, and declined to enter judgment that U.S. Patent No. 10,538,814, U.S. Patent No. 10,557,172, and U.S. Patent No. 10,590,482 are invalid. On January 12, 2021, the court issued an order granting Natera leave to amend its complaint to add Invitae as a co-defendant and plead allegations that ArcherDX and Invitae induce end-users to infringe the patents-in-suit. Natera filed its Second Amended Complaint on the same day and served Invitae on January 15, 2021. The litigations have now been consolidated for all purposes, are ongoing, and trial has been scheduled for May 2022.
If any of ArcherDX’s products or ArcherDX’s use of AMP chemistry is found to infringe any of the Natera Asserted Patents, it could be required to redesign its technology or obtain a license from Natera to continue developing, manufacturing, marketing, selling and commercializing AMP and its products. However, ArcherDX may not be successful in the redesign of its technology or able to obtain any such license on commercially reasonable terms or at all. Even if ArcherDX were able to obtain a license, it could be non-exclusive, thereby giving Natera and other third parties the right to use the same technologies licensed to ArcherDX, and it could require ArcherDX to make substantial licensing, royalty and other payments. ArcherDX also could be forced, including by court order, to permanently cease developing, manufacturing, marketing and commercializing ArcherDX’s products that are found to be infringing. In addition, ArcherDX could be found liable for significant monetary damages, including treble damages and attorneys’ fees, if ArcherDX is found to have willfully infringed any of the Natera Asserted Patents. Even if ArcherDX were ultimately to prevail, litigation with Natera could require us to divert substantial financial and management resources that we would otherwise be able to devote to our business.
We cannot reasonably estimate the final outcome, including any potential liability or any range of potential future charges associated with these litigations. However, any finding of infringement by ArcherDX of any of the Natera Asserted Patents could have a material adverse effect on the business of ArcherDX and the benefits we expected to achieve through our acquisition of ArcherDX, as well as our financial condition and results of operations.
50
Litigation or other proceedings or third-party claims of intellectual property infringement or misappropriation will require us to spend significant time and money, and could in the future prevent us from selling our tests or impact our stock price.
Our commercial success will depend in part on our avoiding infringement of patents and proprietary rights of third parties, including for example the intellectual property rights of competitors. As we continue to commercialize our tests in their current or an updated form, launch different and expanded tests, and enter new markets, competitors might claim that our tests infringe or misappropriate their intellectual property rights as part of business strategies designed to impede our successful commercialization and entry into new markets. Our activities may be subject to claims that we infringe or otherwise violate patents or other intellectual property rights owned or controlled by third parties. For example, as discussed in the preceding risk factor, we are currently engaged in litigation with a competitor of ArcherDX alleging infringement. We cannot assure you that our operations do not, or will not in the future, infringe existing or future patents. We may be unaware of patents that a third party, including for example a competitor in the genetic testing market, might assert are infringed by our business. There may also be patent applications that, if issued as patents, could be asserted against us. Third parties making claims against us for infringement or misappropriation of their intellectual property rights may seek and obtain injunctive or other equitable relief, which could effectively block our ability to perform our tests. Further, if a patent infringement suit were brought against us, we could be forced to stop or delay our development or sales of any tests or other activities that are the subject of such suit. Defense of these claims, regardless of merit, could cause us to incur substantial expenses and be a substantial diversion of our employee resources. Any adverse ruling or perception of an adverse ruling in defending ourselves could have a material adverse impact on our business and stock price. In the event of a successful claim of infringement against us by a third party, we may have to (1) pay substantial damages, possibly including treble damages and attorneys’ fees if we are found to have willfully infringed patents; (2) obtain one or more licenses, which may not be available on commercially reasonable terms (if at all); (3) pay royalties; and/or (4) redesign any infringing tests or other activities, which may be impossible or require substantial time and monetary expenditure, all of which could have a material adverse impact on our cash position and business and financial condition.
If licenses to third-party intellectual property rights are or become required for us to engage in our business, we may be unable to obtain them at a reasonable cost, if at all. Even if such licenses are available, we could incur substantial costs related to royalty payments for licenses obtained from third parties, which could negatively affect our gross margins. Moreover, we could encounter delays in the introduction of tests while we attempt to develop alternatives. Defense of any lawsuit or failure to obtain any of these licenses on favorable terms could prevent us from commercializing tests, which could materially affect our ability to grow and thus adversely affect our business and financial condition.
Developments in patent law could have a negative impact on our business.
Although we view current U.S. Supreme Court precedent to be aligned with our belief that naturally occurring DNA sequences and detection of natural correlations between observed facts (such as patient genetic data) and an understanding of that fact’s implications (such as a patient’s risk of disease associated with certain genetic variations) should not be patentable, it is possible that subsequent determinations by the U.S. Supreme Court or other federal courts could limit, alter or potentially overrule current law. Moreover, from time to time the U.S. Supreme Court, other federal courts, the U.S. Congress or the U.S. Patent and Trademark Office, or USPTO, may change the standards of patentability, and any such changes could run contrary to, or otherwise be inconsistent with, our belief that naturally occurring DNA sequences and detection of natural correlations between observed facts and an understanding of that fact’s implications should not be patentable, which could result in third parties newly claiming that our business practices infringe patents drawn from categories of patents which we currently view to be invalid as directed to unpatentable subject matter. For example, the U.S. Senate Judiciary Committee, Subcommittee on Intellectual Property held hearings in 2019 regarding a legislative proposal that would overrule current U.S. Supreme Court precedent concerning the scope of patentable subject matter. Our President and Chief Executive Officer, Sean George, appeared before this subcommittee. If such proposal were to be formulated as a bill and enacted into law, there could be an increase in third-party claims to patent rights over correlations between patient genetic data and its interpretation and such third parties may assert that our business practices infringe some of those resulting patent rights.
51
Our inability to effectively protect our proprietary technologies, including the confidentiality of our trade secrets, could harm our competitive position.
We currently rely upon trade secret protection and copyright, as well as non-disclosure agreements and invention assignment agreements with our employees, consultants and third parties, and to a limited extent patent protection, to protect our confidential and proprietary information. Although our competitors have utilized and are expected to continue utilizing similar methods and have aggregated and are expected to continue to aggregate similar databases of genetic testing information, our success will depend upon our ability to develop proprietary methods and databases and to defend any advantages afforded to us by such methods and databases relative to our competitors. If we do not protect our intellectual property adequately, competitors may be able to use our methods and databases and thereby erode any competitive advantages we may have.
We will be able to protect our proprietary rights from unauthorized use by third parties only to the extent that our proprietary technologies are covered by valid and enforceable patents or are effectively maintained as trade secrets. In this regard, we have applied, and we intend to continue applying, for patents covering such aspects of our technologies as we deem appropriate. However, we expect that potential patent coverage we may obtain will not be sufficient to prevent substantial competition. In this regard, we believe it is probable that others will independently develop similar or alternative technologies or design around those technologies for which we may obtain patent protection. In addition, any patent applications we file may be challenged and may not result in issued patents or may be invalidated or narrowed in scope after they are issued. Questions as to inventorship or ownership may also arise. Any finding that our patents or applications are unenforceable could harm our ability to prevent others from practicing the related technology, and a finding that others have inventorship or ownership rights to our patents and applications could require us to obtain certain rights to practice related technologies, which may not be available on favorable terms, if at all. If we initiate lawsuits to protect or enforce our patents, or litigate against third-party claims, which would be expensive, and, if we lose, we may lose some of our intellectual property rights. Furthermore, these lawsuits may divert the attention of our management and technical personnel.
We expect to rely primarily upon trade secrets and proprietary know-how protection for our confidential and proprietary information, and we have taken security measures to protect this information. These measures, however, may not provide adequate protection for our trade secrets, know-how or other confidential information. Among other things, we seek to protect our trade secrets and confidential information by entering into confidentiality agreements with employees and consultants. There can be no assurance that any confidentiality agreements that we have with our employees and consultants will provide meaningful protection for our trade secrets and confidential information or will provide adequate remedies in the event of unauthorized use or disclosure of such information. Accordingly, there also can be no assurance that our trade secrets will not otherwise become known or be independently developed by competitors. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret can be difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, trade secrets may be independently developed by others in a manner that could prevent legal recourse by us. If any of our confidential or proprietary information, such as our trade secrets, were to be disclosed or misappropriated, or if any such information was independently developed by a competitor, our competitive position could be harmed.
We may not be able to enforce our intellectual property rights throughout the world.
The laws of some foreign countries do not protect proprietary rights to the same extent as the laws of the United States, and many companies have encountered significant challenges in establishing and enforcing their proprietary rights outside of the United States. These challenges can be caused by the absence of rules and methods for the establishment and enforcement of intellectual property rights outside of the United States. In addition, the legal systems of some countries, particularly developing countries, do not favor the enforcement of patents and other intellectual property protection, especially those relating to healthcare. This could make it difficult for us to stop the infringement of our patents, if obtained, or the misappropriation of our other intellectual property rights. For example, many foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. In addition, many countries limit the enforceability of patents against third parties, including government agencies or government contractors. In these countries, patents may provide limited or no benefit. Patent protection must ultimately be sought on a country-by-country basis, which is an expensive and time-consuming process with uncertain outcomes. Accordingly, we may choose not to seek patent protection in certain countries, and we will not have the benefit of patent protection in such countries. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business. Accordingly, our efforts to protect our intellectual property rights in such countries may be inadequate. In addition, changes in the law and legal decisions by courts in the United States and foreign countries may affect our ability to obtain adequate protection for our technology and the enforcement of intellectual property.
52
Third parties may assert that our employees or consultants have wrongfully used or disclosed confidential information or misappropriated trade secrets.
We employ individuals who were previously employed at universities or genetic testing, diagnostic or other healthcare companies, including our competitors or potential competitors. Although we try to ensure that our employees and consultants do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees or consultants have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of a former employer or other third parties. Further, we may be subject to ownership disputes in the future arising, for example, from conflicting obligations of consultants or others who are involved in developing our intellectual property. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
Risks related to being a public company
We incur increased costs and demands on management as a result of compliance with laws and regulations applicable to public companies, which could harm our operating results.
As a public company, we incur significant legal, accounting and other expenses, including costs associated with public company reporting requirements. In addition, the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, as well as rules implemented by the SEC and the New York Stock Exchange, or NYSE, impose a number of requirements on public companies, including with respect to corporate governance practices. The SEC and other regulators have continued to adopt new rules and regulations and make additional changes to existing regulations that require our compliance. In addition, the Dodd-Frank Wall Street Reform and Consumer Protection Act, or the Dodd-Frank Act, enacted in 2010, includes significant corporate governance and executive-compensation-related provisions. Our management and other personnel need to devote a substantial amount of time to these compliance and disclosure obligations. If these requirements divert the attention of our management and personnel from other aspects of our business concerns, they could have a material adverse effect on our business, financial condition and results of operations. Moreover, these rules and regulations applicable to public companies substantially increase our legal, accounting and financial compliance costs, require that we hire additional personnel and make some activities more time consuming and costly. It may also be more expensive for us to obtain director and officer liability insurance.
If we are unable to maintain effective internal control over financial reporting, investors may lose confidence in the accuracy and completeness of our reported financial information and the market price of our common stock may be negatively affected.
We are required to maintain internal control over financial reporting and to report any material weaknesses in such internal controls. Section 404 of the Sarbanes-Oxley Act requires that we evaluate and determine the effectiveness of our internal control over financial reporting and provide a management report on our internal control over financial reporting. If we have a material weakness in our internal control over financial reporting, we may not detect errors on a timely basis and our financial statements may be materially misstated. We have compiled the system and process documentation necessary to perform the evaluation needed to comply with Section 404 of the Sarbanes-Oxley Act. We need to maintain and enhance these processes and controls as we grow and we have required, and may continue to require, additional personnel and resources to do so.
During the evaluation and testing process, if we identify one or more material weaknesses in our internal controls, our management will be unable to conclude that our internal control over financial reporting is effective. Our independent registered public accounting firm is required to issue an attestation report on the effectiveness of our internal control over financial reporting every fiscal year. Even if our management concludes that our internal control over financial reporting is effective, our independent registered public accounting firm may conclude that there are material weaknesses with respect to our internal controls or the level at which our internal controls are documented, designed, implemented or reviewed.
If we are unable to conclude that our internal control over financial reporting is effective, or if our auditors were to express an adverse opinion on the effectiveness of our internal control over financial reporting because we had one or more material weaknesses, investors could lose confidence in the accuracy and completeness of our financial disclosures, which could cause the price of our common stock to decline. Internal control deficiencies could also result in the restatement of our financial results in the future.
53
Risks related to our indebtedness
The terms of our credit agreement require us to meet certain operating and financial covenants and place restrictions on our operating and financial flexibility. If we raise additional capital through debt financing, the terms of any new debt could further restrict our ability to operate our business.
In October 2020, we entered into a credit agreement with Perceptive Credit Holdings II, LP, pursuant to which we borrowed an aggregate principal amount of $135.0 million, or the 2020 Term Loan. The 2020 Term Loan is secured by a first priority lien on all of our and our subsidiaries’ assets (including our intellectual property) and is guaranteed by our subsidiaries, in each case, excluding certain excluded assets and immaterial subsidiaries. If the 2020 Term Loan is prepaid, we may be required to pay a prepayment fee of up to 6% and a make-whole fee, in each case depending on when the prepayment is made.
The credit agreement restricts our ability to pursue certain mergers, acquisitions, amalgamations or consolidations, other than permitted acquisitions, that we may believe to be in our best interest. In addition, the credit agreement contains financial covenants that require us to maintain a minimum cash balance and minimum quarterly revenue levels. If we default under the credit agreement, the lender will be able to declare all obligations immediately due and payable, including prepayment fees and other obligations. The lender could declare an event of default under the credit agreement upon the occurrence of any event that it interprets as a material adverse change or material adverse effect, each as defined under the credit agreement. Any declaration by the lender of an event of default could significantly harm our business and prospects and could cause the price of our common stock to decline.
If we raise any additional debt financing, the terms of such additional debt could further restrict our operating and financial flexibility.
Servicing our debt requires a significant amount of cash, and we may not have sufficient cash flow from our business to pay our substantial debt.
In September 2019, we issued $350.0 million aggregate principal amount of our Convertible Senior Notes in a private placement and in October 2020 we entered into our credit agreement and borrowed $135.0 million through the 2020 Term Loan.
Our ability to make scheduled payments of the principal of, to pay interest on or to refinance our indebtedness, including the notes, depends on our future performance, which is subject to economic, financial, competitive and other factors beyond our control. Our business may not continue to generate cash flow from operations in the future sufficient to service our debt and make necessary capital expenditures. If we are unable to generate such cash flow, we may be required to adopt one or more alternatives, such as selling assets, restructuring debt or obtaining additional equity capital on terms that may be onerous or highly dilutive. Our ability to refinance our indebtedness will depend on the capital markets and our financial condition at such time. We may not be able to engage in any of these activities or engage in these activities on desirable terms, which could result in a default on our debt obligations.
We may not have the ability to raise the funds necessary to settle conversions of the Convertible Senior Notes in cash or to repurchase the notes upon a fundamental change, and our current credit agreement contains and our future debt may contain limitations on our ability to pay cash upon conversion or repurchase of the notes.
Holders of the notes have the right to require us to repurchase all or any portion of their notes upon the occurrence of a fundamental change at a fundamental change repurchase price equal to 100% of the principal amount of the notes to be repurchased, plus accrued and unpaid interest, if any. In addition, upon conversion of the notes, unless we elect to deliver solely shares of our common stock to settle such conversion (other than paying cash in lieu of delivering any fractional share), we will be required to make cash payments in respect of the notes being converted. However, we only have limited ability to make those cash payments under our credit agreement and, even if the credit agreement limitations are no longer in effect, we may not have enough available cash or be able to obtain financing at the time we are required to make repurchases of notes surrendered therefor or notes being converted. In addition, our ability to repurchase the notes or to pay cash upon conversions of the notes may be limited by law, by regulatory authority or by agreements governing our future indebtedness. Our failure to repurchase notes at a time when the repurchase is required by the indenture or to pay any cash payable on future conversions of the notes as required by the indenture would constitute a default under the indenture. A default under the indenture or the occurrence of the fundamental change itself could also lead to a default under our credit agreement and any agreements governing our future indebtedness. If the repayment of the related indebtedness were to be accelerated after any applicable notice or grace periods, we may not have sufficient funds to repay the indebtedness and to repay or repurchase the notes.
54
The conditional conversion feature of the Convertible Senior Notes, if triggered, may adversely affect our financial condition and operating results.
In the event the conditional conversion feature of the notes is triggered, such as was the case for the quarter ending March 31, 2021, holders of notes will be entitled to convert the notes at any time during specified periods at their option. If one or more holders elect to convert their notes, unless we elect to satisfy our conversion obligation by delivering solely shares of our common stock (other than paying cash in lieu of delivering any fractional share), we would be required to settle a portion or all of our conversion obligation through the payment of cash, which could adversely affect our liquidity. In addition, even if holders do not elect to convert their notes, we could be required under applicable accounting rules to reclassify all or a portion of the outstanding principal of the notes as a current rather than long-term liability, which would result in a material reduction of our net working capital.
The accounting method for convertible debt securities that may be settled in cash, such as the notes, could have a material effect on our reported financial results.
In May 2008, the Financial Accounting Standards Board, or FASB, issued FASB Staff Position No. APB 14-1, Accounting for Convertible Debt Instruments That May Be Settled in Cash Upon Conversion (Including Partial Cash Settlement), which has subsequently been codified as Accounting Standards Codification 470-20, Debt with Conversion and Other Options, or ASC 470-20. Under ASC 470-20, an entity must separately account for the liability and equity components of the convertible debt instruments (such as the notes) that may be settled entirely or partially in cash upon conversion in a manner that reflects the issuer’s economic interest cost. The effect of ASC 470-20 on the accounting for the notes is that the equity component is required to be included in the additional paid-in capital section of stockholders’ equity on our consolidated balance sheet at issuance, and the value of the equity component would be treated as original issue discount for purposes of accounting for the debt component of the notes. As a result, we are required to record a greater amount of non-cash interest expense in current periods presented as a result of the amortization of the discounted carrying value of the notes to their face amount over the term of the notes. We will report larger net losses or lower net income in our financial results because ASC 470-20 will require interest to include both the current period’s amortization of the debt discount and the instrument’s non-convertible coupon interest rate, which could adversely affect our reported or future financial results, the trading price of our common stock and the trading price of the notes.
In addition, under certain circumstances, convertible debt instruments (such as the notes) that may be settled entirely or partly in cash are currently accounted for utilizing the treasury stock method, the effect of which is that the shares issuable upon conversion of the notes are not included in the calculation of diluted net income (loss) per share except to the extent that the conversion value of the notes exceeds their principal amount. Under the treasury stock method, for diluted earnings per share purposes, the transaction is accounted for as if the number of shares of common stock that would be necessary to settle such excess, if we elected to settle such excess in shares, are issued. In August 2020, the FASB amended these accounting standards, effective for fiscal years beginning after December 15, 2021, with early adoption permitted no earlier than fiscal years beginning after December 15, 2020, to eliminate the treasury stock method for convertible instruments and instead require application of the ‘‘if-converted’’ method. Under that method, diluted net income (loss) per share would generally be calculated assuming that all the notes were converted solely into shares of common stock at the beginning of the reporting period, unless the result would be anti-dilutive. The application of the ‘‘if-converted’’ method may reduce our reported diluted net income (or further increase our diluted net loss, as the case may be) per share.
General risk factors
Our stock price is volatile, and you may not be able to sell shares of our common stock at or above the price you paid.
The trading price of our common stock is volatile and could be subject to wide fluctuations in response to various factors, some of which are beyond our control. These factors include:
•actual or anticipated fluctuations in our operating results;
•competition from existing tests or new tests that may emerge;
•announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures, collaborations or capital commitments;
•failure to meet or exceed financial estimates and projections of the investment community or that we provide to the public;
55
•issuance of new or updated research or reports by securities analysts or changed recommendations for our stock;
•our focus on long-term goals over short-term results;
•the level of short interest in our stock, and the effect of short sellers on the price of our stock;
•the timing and magnitude of our investments in the growth of our business;
•actual or anticipated changes in regulatory oversight of our business;
•additions or departures of key management or other personnel;
•disputes or other developments related to our intellectual property or other proprietary rights, including litigation;
•changes in reimbursement by current or potential payers;
•general economic and market conditions; and
•issuances of significant amounts of our common stock.
In addition, the stock market in general, and the market for stock of life sciences companies in particular, has experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of those companies. Broad market and industry factors, including COVID-19, may seriously affect the market price of our common stock, regardless of our actual operating performance. In addition, in the past, following periods of volatility in the overall market and the market price of a particular company’s securities, securities class action litigation has often been instituted against these companies. This litigation, if instituted against us, could result in substantial costs and a diversion of our management’s attention and resources.
If securities or industry analysts issue an adverse opinion regarding our stock or do not publish research or reports about our company, our stock price and trading volume could decline.
The trading market for our common stock will depend in part on the research and reports that equity research analysts publish about us and our business. We do not control these analysts or the content and opinions included in their reports. Securities analysts may elect not to provide research coverage of our company and such lack of research coverage may adversely affect the market price of our common stock. The price of our common stock could also decline if one or more equity research analysts downgrade our common stock, change their price targets, issue other unfavorable commentary or cease publishing reports about us or our business. If one or more equity research analysts cease coverage of our company, we could lose visibility in the market, which in turn could cause our stock price to decline.
We have never paid dividends on our capital stock, and we do not anticipate paying dividends in the foreseeable future.
We have never paid dividends on any of our capital stock and currently intend to retain any future earnings to fund the growth of our business. In addition, the terms of our credit agreement restrict our ability to pay dividends. Any determination to pay dividends in the future will be at the discretion of our board of directors and will depend on our financial condition, operating results, capital requirements, general business conditions and other factors that our board of directors may deem relevant. As a result, capital appreciation, if any, of our common stock will be the sole source of gain for the foreseeable future.
Anti-takeover provisions in our charter documents and under Delaware law could discourage, delay or prevent a change in control and may affect the trading price of our common stock.
Provisions in our restated certificate of incorporation and our amended and restated bylaws may have the effect of delaying or preventing a change of control or changes in our management. Our restated certificate of incorporation and amended and restated bylaws include provisions that:
•authorize our board of directors to issue, without further action by the stockholders, up to 20,000,000 shares of undesignated preferred stock;
•require that any action to be taken by our stockholders be effected at a duly called annual or special meeting and not by written consent;
•specify that special meetings of our stockholders can be called only by our board of directors, our chairman of the board or our chief executive officer;
56
•establish an advance notice procedure for stockholder approvals to be brought before an annual meeting of our stockholders, including proposed nominations of persons for election to our board of directors;
•establish that our board of directors is divided into three classes, Class I, Class II and Class III, with each class serving staggered terms;
•provide that our directors may be removed only for cause;
•provide that vacancies on our board of directors may, except as otherwise required by law, be filled only by a majority of directors then in office, even if less than a quorum; and
•require a super-majority of votes to amend certain of the above-mentioned provisions as well as to amend our bylaws generally.
In addition, we are subject to the provisions of Section 203 of the Delaware General Corporation Law regulating corporate takeovers. Section 203 generally prohibits us from engaging in a business combination with an interested stockholder subject to certain exceptions.
Our certificate of incorporation designates the Court of Chancery of the State of Delaware as the sole and exclusive forum for certain types of actions and proceedings that may be initiated by our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or other employees.
Our certificate of incorporation provides that, unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware shall be the sole and exclusive forum for:
•any derivative action or proceeding brought on our behalf;
•any action asserting a claim of breach of a fiduciary duty owed by any of our directors, officers, or other employees to us or our stockholders;
•any action asserting a claim arising pursuant to any provision of the Delaware General Corporation Law; or
•any action asserting a claim against us governed by the internal affairs doctrine.
Section 27 of the Exchange Act creates exclusive federal jurisdiction over all suits brought to enforce any duty or liability created by the Exchange Act or the rules and regulations thereunder. As a result, the exclusive forum provision in our certificate of incorporation will not apply to suits brought to enforce any duty or liability created by the Exchange Act or any other claim for which the federal courts have exclusive jurisdiction. Section 22 of the Securities Act creates concurrent jurisdiction for federal and state courts over all suits brought to enforce any duty or liability created by the Securities Act or the rules and regulations thereunder. As a result, the exclusive forum provision in our certificate of incorporation will not apply to suits brought to enforce any duty or liability created by the Securities Act or any other claim for which the federal and state courts have concurrent jurisdiction.
Any person or entity purchasing or otherwise acquiring any interest in shares of our capital stock shall be deemed to have notice of and consented to the provisions of our certificate of incorporation described above. This choice of forum provision may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits against us and our directors, officers and other employees. Alternatively, if a court were to find these provisions of our certificate of incorporation inapplicable to, or unenforceable in respect of, one or more of the specified types of actions or proceedings, we may incur additional costs associated with resolving such matters in other jurisdictions, which could adversely affect our business, financial condition or results of operations.
57
Sales of a substantial number of shares of our common stock in the public market could cause our stock price to fall.
Sales of a substantial number of shares of our common stock in the public market or the perception that these sales might occur could depress the market price of our common stock and could impair our ability to raise capital through the sale of additional equity securities. As of December 31, 2020, we had outstanding 185.9 million shares of our common stock, options to purchase 4.8 million shares of our common stock (of which 4.4 million were exercisable as of that date), outstanding restricted stock units, or RSUs, representing 6.6 million shares of our common stock (which includes an estimated number of RSUs, subject to certain employee’s continued service with us, or Time-based RSUs, and RSUs that are performance based RSUs, or PRSUs, granted in connection with an acquisition), outstanding Series A convertible preferred stock convertible into 0.1 million shares of our common stock and warrants to purchase 0.2 million shares of our common stock. The foregoing does not include shares that may be issuable in connection with indemnification hold-backs and contingent consideration related to our acquisitions other than ArcherDX, and up to 22.0 million shares which may be issuable upon the achievement of certain milestones related to our acquisition of ArcherDX, inducement awards issued in connection with an acquisition, or shares that may be issuable in the future in connection with the convertible senior notes. Also not included are the shares issued or issuable in connection with acquisitions after December 31, 2020, including approximately 1.4 million shares of our common stock that we will register for resale following the filing of this Report. The sale or the availability for sale of a large number of shares of our common stock in the public market could cause the price of our common stock to decline.
58
ITEM 1B. Unresolved Staff Comments.
None.
ITEM 2. Properties.
Our headquarters and main production facility is located in San Francisco, California, where we currently lease and occupy approximately 103,000 square feet of laboratory and office space. The lease for this facility expires in July 2026 and we may renew the lease for an additional ten years.
We also lease approximately 330,000 square feet of additional office and laboratory space domestically in California, Colorado, Massachusetts, New York and Washington, and internationally in Australia and Israel.
We believe that our facilities are adequate for our current needs and that additional space will be available on commercially reasonable terms if required.
ITEM 3. Legal Proceedings.
For a discussion of legal matters as of December 31, 2020, see Note 8, “Commitments and contingencies” in Notes to Consolidated Financial Statements in Part II, Item 8 of this report, which is incorporated into this item by reference.
ITEM 4. Mine Safety Disclosure.
Not applicable.
59
PART II
ITEM 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Our common stock has been publicly traded on the New York Stock Exchange under the symbol “NVTA” since February 12, 2015. Prior to that time, there was no public market for our common stock.
As of February 19, 2021, there were 247 stockholders of record of our common stock. The actual number of stockholders is greater than this number of record holders and includes stockholders who are beneficial owners but whose shares are held in street name by brokers and other nominees.
We have never declared or paid any cash dividends on our capital stock. We currently intend to retain any future earnings and do not expect to pay any dividends in the foreseeable future. In addition, the terms of the credit agreement restrict our ability to pay dividends. Any determination to pay dividends in the future will be at the discretion of our board of directors and will depend on our financial condition, operating results, capital requirements and general business conditions and other factors that our board of directors may deem relevant.
Stock performance graph
The following information shall not be deemed to be soliciting material or to be filed with the SEC, or subject to Regulations 14A or 14C under the Securities Exchange Act of 1934, or Exchange Act, or to the liabilities of Section 18 of the Exchange Act nor shall such information be incorporated by reference into any future filing under the Securities Act or the Exchange Act, except to the extent that we specifically incorporate it by reference into such filing.
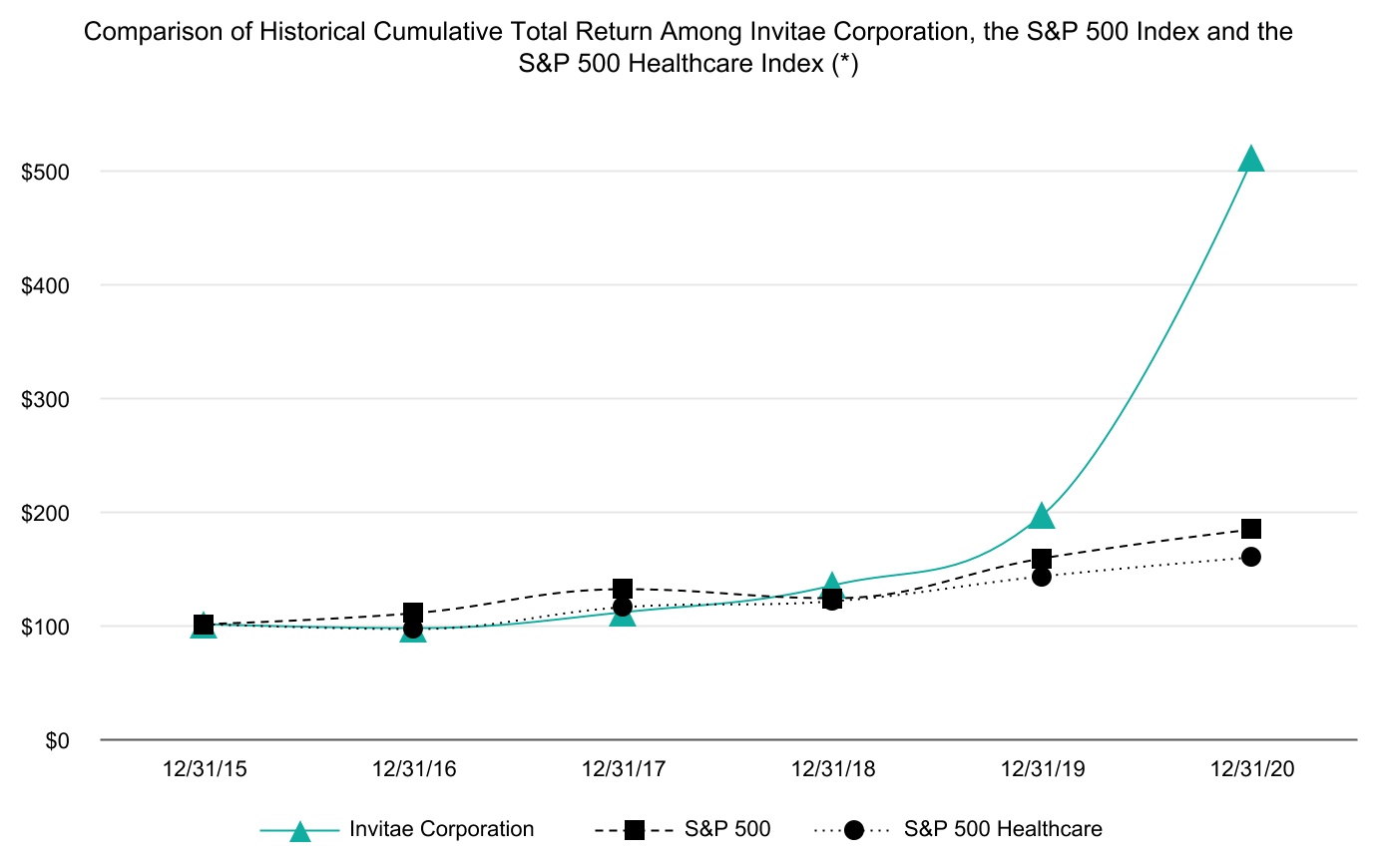
_________________________________________________________________
(*)The above graph shows the cumulative total stockholder return of an investment of $100 in cash on December 31, 2015 through December 31, 2020 for: (i) our common stock; (ii) the S&P 500 Index; and (iii) the S&P 500 Healthcare Index. All values assume reinvestment of the full amount of all dividends. The comparisons in the table are not intended to be forecasts or indicative of future stockholder returns.
60
| 12/31/2015 | 12/31/2016 | 12/31/2017 | 12/31/2018 | 12/31/2019 | 12/31/2020 | ||||||||||||||||||||||||||||||||||||
| Invitae Corporation | $ | 100.00 | $ | 96.71 | $ | 110.60 | $ | 134.71 | $ | 196.47 | $ | 509.26 | |||||||||||||||||||||||||||||
| S&P 500 | $ | 100.00 | $ | 109.54 | $ | 130.81 | $ | 122.65 | $ | 158.07 | $ | 183.77 | |||||||||||||||||||||||||||||
| S&P 500 Healthcare Index | $ | 100.00 | $ | 95.64 | $ | 114.77 | $ | 120.16 | $ | 142.60 | $ | 158.90 | |||||||||||||||||||||||||||||
ITEM 6. Selected Financial Data.
The information set forth below should be read together with “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our audited consolidated financial statements and related notes included elsewhere in this report. The selected consolidated balance sheet data at December 31, 2020 and 2019 and the selected consolidated statements of operations data for each of the years ended December 31, 2020, 2019, and 2018 have been derived from our audited consolidated financial statements that are included elsewhere in this report. The selected consolidated balance sheet data at December 31, 2018, 2017 and 2016 and the selected consolidated statement of operations data for the years ended December 31, 2017 and 2016 have been derived from our audited consolidated financial statements not included in this report. Historical results are not necessarily indicative of results to be expected in any future period.
| Year Ended December 31, | |||||||||||||||||||||||||||||
| 2020 | (1) | 2019 | (1) | 2018 | (3) | 2017 | (1) | 2016 | |||||||||||||||||||||
| (In thousands, except per share data) | |||||||||||||||||||||||||||||
| Consolidated Statements of Operations Data: | |||||||||||||||||||||||||||||
| Test revenue | $ | 272,310 | $ | 212,473 | $ | 144,560 | $ | 65,169 | $ | 24,840 | |||||||||||||||||||
| Other revenue | 7,288 | 4,351 | 3,139 | 3,052 | 208 | ||||||||||||||||||||||||
| Total revenue | 279,598 | 216,824 | 147,699 | 68,221 | 25,048 | ||||||||||||||||||||||||
Cost of revenue (4) | 198,275 | 118,103 | 80,105 | 50,142 | 27,878 | ||||||||||||||||||||||||
Research and development (4) | 240,605 | 141,526 | 63,496 | 46,469 | 44,630 | ||||||||||||||||||||||||
Selling and marketing (4) | 168,317 | 122,237 | 74,428 | 53,417 | 28,638 | ||||||||||||||||||||||||
General and administrative (4) | 324,573 | 79,070 | 52,227 | 39,472 | 24,085 | ||||||||||||||||||||||||
| Loss from operations | (652,172) | (244,112) | (122,557) | (121,279) | (100,183) | ||||||||||||||||||||||||
| Other income (expense), net | (32,332) | (3,891) | (2,568) | (303) | 348 | ||||||||||||||||||||||||
| Interest expense | (29,766) | (12,412) | (7,030) | (3,654) | (421) | ||||||||||||||||||||||||
| Net loss before taxes | (714,270) | (260,415) | (132,155) | (125,236) | (100,256) | ||||||||||||||||||||||||
| Income tax benefit | (112,100) | (18,450) | (2,800) | (1,856) | — | ||||||||||||||||||||||||
| Net loss | $ | (602,170) | $ | (241,965) | $ | (129,355) | $ | (123,380) | $ | (100,256) | |||||||||||||||||||
Net loss per share, basic and diluted (5) | $ | (4.47) | $ | (2.66) | $ | (1.94) | $ | (2.65) | $ | (3.02) | |||||||||||||||||||
| Shares used in computing net loss per share, basic and diluted | 134,587 | 90,859 | 66,747 | 46,512 | 33,176 | ||||||||||||||||||||||||
61
| As of December 31, | |||||||||||||||||||||||||||||
| 2020 | (1) | 2019 | (1,2) | 2018 | (3) | 2017 | (1) | 2016 | |||||||||||||||||||||
| (In thousands) | |||||||||||||||||||||||||||||
| Consolidated Balance Sheet Data: | |||||||||||||||||||||||||||||
| Cash and cash equivalents | $ | 124,794 | $ | 151,389 | $ | 112,158 | $ | 12,053 | $ | 66,825 | |||||||||||||||||||
| Marketable securities | 229,186 | 240,436 | 13,727 | 52,607 | 25,798 | ||||||||||||||||||||||||
| Working capital | 332,187 | 360,538 | 129,127 | 53,294 | 87,047 | ||||||||||||||||||||||||
| Total assets | 3,430,485 | 781,601 | 282,959 | 211,078 | 130,651 | ||||||||||||||||||||||||
| Debt | 104,449 | — | 74,477 | 39,084 | 12,102 | ||||||||||||||||||||||||
| Convertible senior notes, net | 283,724 | 268,755 | — | — | — | ||||||||||||||||||||||||
| Total liabilities | 1,454,192 | 401,961 | 121,120 | 89,284 | 31,577 | ||||||||||||||||||||||||
| Accumulated deficit | (1,360,847) | (758,677) | (516,712) | (398,598) | (275,218) | ||||||||||||||||||||||||
| Total stockholders' equity | 1,976,293 | 379,640 | 161,839 | 121,794 | 99,074 | ||||||||||||||||||||||||
___________________________________________________________________
(1)In 2020 we completed the acquisition of four businesses, including ArcherDX, in 2019 we completed the acquisition of three businesses, and in 2017 we completed the acquisition of four businesses, all of which are included in our selected consolidated financial data as of the applicable acquisition date.
(2)On January 1, 2019, we adopted Accounting Standards Codification, or ASC, Topic 842 using the modified retrospective transition method as of the adoption date which required the recognition of operating lease right-of-use assets and operating lease liabilities to be recognized on our consolidated balance sheets. Prior period amounts are presented as originally reported based upon the accounting standards in effect for those periods.
(3)On January 1, 2018, we adopted ASC Topic 606 using the modified retrospective transition method. Prior period amounts are presented as originally reported based upon the accounting standards in effect for those periods.
(4)Includes employee stock-based compensation as follows (in thousands):
| Year Ended December 31, | |||||||||||||||||||||||||||||
| 2020 | 2019 | 2018 | 2017 | 2016 | |||||||||||||||||||||||||
| Cost of revenue | $ | 8,713 | $ | 4,563 | $ | 2,960 | $ | 2,093 | $ | 1,353 | |||||||||||||||||||
| Research and development | 91,762 | 52,450 | 7,017 | 6,158 | 4,976 | ||||||||||||||||||||||||
| Selling and marketing | 14,418 | 7,641 | 4,887 | 3,956 | 1,709 | ||||||||||||||||||||||||
| General and administrative | 43,854 | 11,294 | 5,986 | 7,014 | 2,661 | ||||||||||||||||||||||||
| Total stock-based compensation | $ | 158,747 | $ | 75,948 | $ | 20,850 | $ | 19,221 | $ | 10,699 | |||||||||||||||||||
See Note 4, "Business combinations," and Note 10, "Stock incentive plans," in our audited consolidated financial statements included elsewhere in this report for further information regarding our stock-based compensation.
(5)See Note 2, "Summary of significant accounting policies," and Note 12, "Net loss per share," in our audited consolidated financial statements included elsewhere in this report for an explanation of the calculations of our basic and diluted net loss per share.
62
ITEM 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
The following discussion of our financial condition and results of operations should be read in conjunction with our financial statements and the related notes included in Item 8 of this report. Historic results are not necessarily indicative of future results.
Business overview
We offer high-quality, comprehensive, affordable genetic testing across multiple clinical areas, including hereditary cancer, cardiology, neurology, pediatrics, personalized oncology, metabolic conditions and rare diseases. To augment our offering and realize our mission, we have acquired multiple assets including four businesses in 2020, which expanded our suite of genome management offerings and established a broader entry into oncology therapy selection and personalized cancer monitoring.
In October 2020, we completed the acquisition of ArcherDX, Inc. (“ArcherDX”), a genomics company democratizing precision oncology by offering a suite of products and services that are accurate, personal, actionable and easy to use in local settings, thereby empowering clinicians to control the sample, data, patient care and economics. ArcherDX’s development platform, including its proprietary Anchored Multiplex PCR, or AMP, chemistry at the core, is enabling clinical tests and services that allow for therapy selection and cancer monitoring in community locations for the first time at scale. In addition, applying these assets via biopharma partnerships may enable more efficient development of new cancer therapies, companion diagnostics and result in more productive clinical trial processes.
We have experienced rapid growth. For the years ended December 31, 2020, 2019 and 2018, our revenue was $279.6 million, $216.8 million and $147.7 million, respectively and we incurred net losses of $602.2 million, $242.0 million and $129.4 million, respectively. At December 31, 2020, our accumulated deficit was $1.4 billion. To meet the demands of scaling our business, we increased our number of employees to approximately 2,100 at December 31, 2020 from approximately 1,300 at December 31, 2019. Our sales force grew to approximately 300 at December 31, 2020 from approximately 230 at December 31, 2019. We expect headcount will continue to increase as we add staff to support anticipated growth.
Sales of our tests have grown significantly. In 2020, 2019 and 2018, we generated 659,000, 469,000 and 292,000 billable units, respectively. We calculate volume using billable units, which are billable events that include individual test reports released and individual reactions shipped. We refer to the set of reagents needed to perform an NGS test as a "reaction." Through December 31, 2020, 46% of the billable units we performed have been billable to patients, biopharma partners and other business-to-business customers (e.g., hospitals, clinics, medical centers), and the remainder have been billable to third-party payers. Many of the gene tests on our assays are tests for which insurers reimburse. However, when we do not have reimbursement policies or contracts with private insurers, our claims for reimbursement may be denied upon submission, and we must appeal the claims. The appeals process is time consuming and expensive, and may not result in payment. Even if we are successful in achieving reimbursement, we may be paid at lower rates than if we were under contract with the third-party payer. When there is not a contracted rate for reimbursement, there is typically a greater payment requirement from the patient that may result in further delay in payment for these tests.
We expect to incur operating losses for the near term as we continue to invest in our business to achieve our revenue growth objectives, including expansion of our platform to capture the broad potential of genetics across healthcare, and may need to raise additional capital in order to fund our operations. If we are unable to achieve these objectives and successfully manage our costs, we may not be able to achieve profitability in the near term or at all.
We believe that the keys to our future growth will be to increase billable volume, achieve broad reimbursement coverage for our tests from third-party payers, drive down the price for genetic analysis and interpretation, steadily increase the amount of genetic content we offer, consistently improve the client experience, drive physician and patient utilization of our website for ordering and delivery of results and increase the number of strategic partners working with us to add value for our clients. We also believe that providing a unique genetic testing platform that is agnostic to stage of life or disease category will deliver unique benefits to customers, payers and other institutions that are seeking to make genetic information a standard element of healthcare decisions in the future.
63
Impact of COVID-19
Our test volumes decreased significantly in the second half of March 2020 as compared to the first few months of 2020 as a result of COVID-19 and related limitations and priorities across the healthcare system. Our daily volumes have consistently increased from the low in March 2020, although we are currently still experiencing changes in product mix due to the impact of COVID-19. COVID-19 could have a material impact on our financial results for the foreseeable future, particularly on product mix and as a result, the revenue we recognize. We have reviewed and adjusted for the impact of COVID-19 on our estimates related to revenue recognition and expected credit losses.
In response to the pandemic we have implemented measures to protect the health of all of our employees during this time with additional measures in place to better protect our on-site lab production and support teams. Our production facilities currently remain fully operational. While we have not experienced significant disruption in our supply chain, over the past several months, we have experienced supply delays as a result of the COVID-19 pandemic and have also had to obtain supplies from new suppliers. Although we do not yet know the full impact COVID-19 will have on our supply chain, we have increased our inventory on hand to respond to potential future disruptions that may occur.
Many announced healthcare guidelines call for a shift of regular physician visits and healthcare delivery activities to remote/telehealth formats. This is particularly important for patients who, despite the fall-out from COVID-19, continue to be diagnosed with critical diseases, like cancer, and for women who are pregnant or are trying to conceive. We believe our investments in new access platforms and technologies will position us well to provide a range of testing to clinicians and patients using a “clinical care from afar” model. An example is our rollout in April 2020 of our Gia telehealth platform, which expands access to remote interaction between patients and clinicians as well as direct ordering of genetic tests. Such access helped to counteract some of the adverse in-office impacts of COVID-19, allowing continuation of key testing categories in a safe environment.
Given the unknown duration and extent of COVID-19’s impact on our business, and the healthcare system in general, we are adapting our spending and investment levels to evolving market conditions, including focusing commercial execution on workflows that support remote ordering, online support and telehealth. Approximately 8% of our workforce as of March 2020 was impacted by a reduction in force in April 2020 in an initiative to manage costs and cash burn, which resulted in one-time costs in the second quarter of 2020 of $3.8 million. In addition, effective May 2020, we reduced the salaries of our named executive officers by approximately 20%, which reductions ceased as of January 2021.
On March 27, 2020, the Coronavirus Aid, Relief, and Economic Security Act, or the CARES Act, was signed into law which was a stimulus bill intended to bolster the economy, among other things, and provide assistance to qualifying businesses and individuals. The CARES Act included an infusion of funds into the healthcare system, and in April 2020, we received $3.8 million as a part of this initiative. This payment was recognized as other income (expense), net in our consolidated statement of operations during the year ended December 31, 2020. We also received $2.3 million during January 2021 which we recognized as other income (expense), net during the three months ended March 31, 2021. At this time, we are not certain of the availability, extent or impact of any future relief provided under the CARES Act.
Factors affecting our performance
Number of billable units
Our centralized test revenue is tied to the number of tests which we bill third-party payers, biopharmaceutical partners, other business-to-business customers (e.g., hospitals, clinics, medical centers), or patients. Our decentralized product revenue is tied to the number of individual reactions we ship biopharma partners and other business-to-business customers. We refer to the set of reagents needed to perform an NGS test as a "reaction," and we refer to billable events that include individual test reports released and individual reactions shipped as billable units. We typically bill for our services following delivery of the billable report derived from testing samples and interpreting the results. For units manufactured for use by customers in distributed facilities, we typically bill customers upon shipment of those units. Test orders are placed under signed requisitions or contractual agreements, as we often enter into contracts with biopharmaceutical partners, other business-to-business customers (e.g., hospitals, clinics, medical centers) and insurance companies that include pricing provisions under which such tests are billed. We incur the expenses associated with a unit in the period in which the unit is processed regardless of when payment is received with respect to that unit. We believe the number of billable units in any period is an important indicator of the growth in our testing business, and with time, this will translate into the number of customers accessing our platform.
64
Number and size of research and commercial partnerships
Pharma development services revenue, which we recognize within other revenue in our consolidated statements of operations, is generated primarily from services provided to biopharmaceutical companies and other partners and is related to companion diagnostic development, clinical research, and clinical trial services across the research, development, and commercialization phases of collaborations. The result of these relationships may include the development of new targeted companion diagnostics, which underscore and expand the need for genetic testing and in some cases may lead to intellectual property and/or revenue sharing opportunities with third-party partners.
In addition to research partnerships, we also seek to grow the number of biopharmaceutical partners and other business-to-business customers (e.g., hospitals, clinics, medical centers) for whom we provide testing technologies, analysis, supplies and expertise to institutions that provide independent testing services to customers in their respective regions.
Success obtaining and maintaining reimbursement
Our ability to increase volume and revenue will depend in part on our success achieving broad reimbursement coverage and laboratory service contracts for our tests from third-party payers and agreements with institutions and partners. Reimbursement may depend on a number of factors, including a payer’s determination that a test is appropriate, medically necessary and cost-effective, as well as whether we are in contract, where we get paid more consistently and at higher rates. Because each payer makes its own decision as to whether to establish a policy or enter into a contract to reimburse for our testing services and specific tests, seeking these approvals is a time-consuming and costly process. In addition, clinicians and patients may decide not to order our tests if the cost of the test is not covered by insurance. Because we require an ordering physician to requisition a test, our revenue growth also depends on our ability to successfully promote the adoption of our testing services and expand our base of ordering clinicians. We believe that establishing coverage and obtaining contracts from third-party payers is an important factor in gaining adoption by ordering clinicians. Our arrangements for laboratory services with payers cover approximately 315 million lives, comprised of Medicare, all national commercial health plans, and Medicaid in most states, including California (Medi-Cal), our home state.
Ability to lower the costs associated with performing our tests
Reducing the costs associated with performing our genetic tests is both a focus and a strategic objective of ours. Over the long term, we will need to reduce the cost of raw materials by improving the output efficiency of our assays and laboratory processes, modifying our platform-agnostic assays and laboratory processes to use materials and technologies that provide equal or greater quality at lower cost, improve how we manage our materials, port some tests onto a next generation sequencing platform and negotiate favorable terms for our materials purchases. Our acquisition of Singular Bio, Inc. is a component of this objective and we expect the technology acquired in this transaction, once developed, to help decrease the costs associated with our NIPS offering. We also intend to continue to design and implement hardware and software tools that are designed to reduce personnel-related costs for both laboratory and clinical operations/medical interpretation by increasing personnel efficiency and thus lowering labor costs per test. Finally, we plan to reduce the cost of providing test equipment and software to laboratories and other facilities in the U.S. and internationally. Those efforts are designed to enable more rapid expansion of genetic testing and patient access, enlarging our geographic footprint outside the U.S. while achieving lower costs.
Ability to expand our genetic content and create new pathways to test
Our focus on reducing the average cost per test will have a countervailing force — increasing the number of tests we offer, the content of each test and the means to connect our testing services with patients and physicians. We intend to continue to expand our test menus by steadily releasing additional genetic content for the same or lower prices per test, ultimately leading to affordable whole genome services. The breadth and flexibility of our offering will be a critical factor in our ability to address new markets, including internationally, for genetic testing services. Both of these, in conjunction with our continued focus on strategic partnerships, will be important to our ability to continue to grow the volume of billable tests we deliver. We have and will continue to identify new ways to connect our testing services and information to patients. These include direct patient outreach and ordering capacity, the use of automated assistants for physician customers to assist with the ease of ordering and processing genetic tests and programs designed to reach underserved patient populations with genetic testing.
65
Investment in our business and timing of expenses
We plan to continue to invest in our genetic testing and information management business. We deploy state-of-the-art technologies in our genetic testing services, and we intend to continue to scale our infrastructure, including our testing capacity and information systems. We also expect to incur software development costs as we seek to further automate our laboratory processes and our genetic interpretation and report sign-out procedures, scale our customer service capabilities to improve our customers' experience, and expand the functionality of our website. We also expect to incur costs as we seek to provide the testing equipment and software necessary to enable decentralized genetic and genomic testing in the U.S. and internationally. We will incur costs related to marketing and branding as we spread our initiatives beyond our current customer base and focus on providing access to customers through our website. We plan to hire additional personnel as necessary to support anticipated growth, including software engineers, sales and marketing personnel, billing personnel, research and development personnel, medical specialists, biostatisticians and geneticists. We will also incur additional costs related to the expansion of our production facilities to accommodate growth and as we expand internationally. In addition, we expect to incur ongoing expenses as a result of operating as a public company. The expenses we incur may vary significantly by quarter as we focus on building out different aspects of our business.
How we recognize revenue
We generally recognize revenue on an accrual basis, which is when a customer obtains control of the promised goods or services, typically a test report. Accrual amounts recognized are based on estimates of the consideration that we expect to receive, and such estimates are adjusted and subsequently recorded until fully settled. Changes to such estimates may increase or decrease revenue recognized in future periods. Revenue from our tests may not be equal to billed amounts due to a number of factors, including differences in reimbursement rates, the amounts of patient payments, the existence of secondary payers and claim denials. Some test orders are placed under signed requisitions or contractual agreements, and we often enter into contracts with biopharmaceutical partners, other business-to-business customers (e.g., hospitals, clinics, medical centers) and insurance companies that include pricing provisions under which such tests are billed.
Pharma development service revenues are generated primarily from custom assay design services, sample processing activities and consultative inputs, which is separate from revenue generated by any related or unrelated product component. Revenue is recognized as samples are processed or scope of work is completed based on contracted agreements with those biopharmaceutical customer companies.
Under these collaborations we also generate revenue from achievement of milestones, provision of on-going support, and related pass-through costs and fees. We generally have distinct performance obligations for development milestones related to our development of a companion diagnostic device. We use a cost plus a margin approach to estimate the standalone value of our companion diagnostic development service performance obligations. Revenue is recognized over time using input or output methods based on our assessments of performance completed to date toward each milestone.
Financial overview
Revenue
We primarily generate revenue from testing services and sales of distributed precision oncology products. Customers are typically billed upon delivery of test results or shipment of products. We also generate revenue from development agreements with biopharmaceutical customers. Our ability to increase our revenue will depend on our ability to increase our market penetration, obtain FDA and other international regulatory authority approvals on future products and services offerings, obtain contracted reimbursement coverage from third-party payers, and grow our relationships with biopharmaceutical customers.
66
Cost of revenue
Cost of revenue reflects the aggregate costs incurred in delivering our products and services and includes expenses for materials and supplies, personnel-related costs, freight, costs for lab services and clinical trial support, equipment and infrastructure expenses and allocated overhead including rent, information technology, equipment depreciation, amortization of acquired intangibles, and utilities. We expect cost of revenue to generally increase in line with the increase in billable volume, however, we expect a future increase in amortization of acquired intangible assets that is not dependent on billed volume. We anticipate our cost per unit for existing tests will generally decrease over time due to the efficiencies we expect to gain as volume increases and from automation and other cost reductions. These reductions in cost per unit will likely be offset by new offerings which often have a higher costs per unit during the introductory phases before we are able to gain efficiencies. The cost per unit may fluctuate significantly from quarter to quarter.
Operating expenses
Our operating expenses are classified into three categories: research and development, selling and marketing, and general and administrative. For each category, the largest component is generally personnel-related costs, which include salaries, employee benefit costs, bonuses, commissions, as applicable, and stock-based compensation expense.
Research and development
Research and development expenses represent costs incurred to develop our technology and future offerings. These costs are principally for process development associated with our efforts to expand the number of genes we can evaluate, our efforts to lower the costs per unit and our development of new products to expand our platofrm. In addition, we incur process development costs to further develop the software we use to operate our laboratories, analyze generated data, process customer orders, validate clinical activities, enable ease of customer ordering, deliver reports and automate our business processes. These costs consist of personnel-related costs, including stock-based compensation, laboratory supplies and equipment expenses, consulting costs, amortization of acquired intangible assets, and allocated overhead including rent, information technology, equipment depreciation and utilities.
We expense all research and development costs in the periods in which they are incurred. We expect our research and development expenses to increase as we continue our efforts to develop additional offerings, make investments to reduce costs, streamline our technology to provide patients access to testing, scale our business domestically and internationally and acquire and integrate new technologies, including those acquired through our acquisition of ArcherDX.
During June 2019 through our acquisition of Singular Bio, we recognized $30.0 million of in-process R&D technology using an income approach. This technology is estimated to be developed in 2021 with significant development costs incurred during the second half of 2019 through 2020 and expected through development completion. If not completed timely, the ability to lower the cost of our NIPS offering may be delayed. During October 2020 through our acquisition of ArcherDX, we recognized $512.4 million of in-process R&D technology for two assets representing STRATAFIDE and Personalized Cancer Monitoring, or PCM, technologies, both using an income approach. We estimate these technologies to be developed in the next few years with significant development costs through completion.
Selling and marketing
Selling and marketing expenses consist of personnel-related costs, including commissions, client service expenses, advertising and marketing expenses, educational and promotional expenses, market research and analysis, and allocated overhead including rent, information technology, equipment depreciation, amortization of acquired intangibles, and utilities. We expect our selling and marketing expenses to increase as we continue to build our brand and focus on advertising our products and services.
67
General and administrative
General and administrative expenses include executive, finance and accounting, billing and collections, legal and human resources functions as well as other administrative costs. These expenses include personnel-related costs; audit, accounting and legal expenses; consulting costs; allocated overhead including rent, information technology, equipment depreciation, and utilities; costs incurred in relation to our co-development agreements; changes in the fair value of contingent consideration related to our acquisitions; and post-combination expenses incurred in relation to companies we acquire. We expect our general and administrative expenses to increase as we support continued growth of operations.
Other expense, net
Other expense, net, primarily consists of adjustments to the fair value of our stock payable liabilities arising from business combinations, and we expect it to fluctuate significantly from period to period due to the volatility of our common stock. Other expense, net also includes income generated from our cash equivalents and marketable securities and amounts received under the CARES Act.
Interest expense
Interest expense is primarily attributable to interest incurred related to our debt financings and finance leases. See Note 8, “Commitments and contingencies” in Notes to Consolidated Financial Statements in Part II, Item 8 of this Annual Report for more details.
Income tax benefit
Since we generally establish a full valuation allowance against our deferred tax balances, our income tax benefit primarily consists of tax impacts of our deferred income tax assessments resulting from our acquisitions.
Critical accounting policies and estimates
Management’s discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles, or U.S. GAAP. The preparation of these financial statements requires us to make judgments, estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported revenue generated and expenses incurred during the reporting periods. We evaluate our estimates on an ongoing basis. Our estimates are based on current facts, our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions and any such differences may be material. We believe that the accounting policies discussed below are critical to understanding our historical and future performance, as these policies relate to the more significant areas involving management’s judgments and estimates.
Revenue recognition
We recognize revenue when control of the promised goods or services is transferred to the customer in an amount that reflects the consideration we expect to be entitled to in exchange for those goods or services. All revenues are generated from contracts with customers. We utilize the following practical expedients and exemptions:
•Costs to obtain or fulfill a contract are expensed when incurred because the amortization period would have been one year or less, and
•No adjustments to promised consideration were made for financing as we expect, at contract inception, that the period between the transfer of a promised good or service and when the customer pays for that good or service will be one year or less.
Test revenue
Test revenue is comprised of testing services and sales of distributed precision oncology products.
68
The majority of our test revenue is generated from genetic testing, in addition to somatic testing for therapy selection and personalized cancer monitoring. These testing services provide analysis and associated interpretation of the sequencing of parts of the genome. Test orders are placed under signed requisitions or contractual agreements, and we often enter into contracts with biopharmaceutical partners, other business-to-business customers (e.g., hospitals, clinics, medical centers) and insurance companies that include pricing provisions under which such tests are billed. Billing terms are generally net 30 to 60 days.
While the transaction price of diagnostic tests is originally established either via contract or pursuant to our standard list price, we often provide concessions for tests billed to insurance carriers, and therefore the transaction price for patient insurance-billed tests is considered to be variable and revenue is recognized based on an estimate of the consideration to which we will be entitled at an amount for which it is probable that a reversal of cumulative consideration will not occur. Making these estimates requires significant judgments based upon such factors as length of payer relationship, historical payment patterns, changes in contract provisions and insurance reimbursement policies. These judgments are reviewed each reporting period and updated as necessary.
We look to transfer of control in assessing timing of recognition of revenue in connection with each performance obligation. In general, revenue in connection with the service portion of our diagnostic tests is recognized upon delivery of the underlying clinical report or when the report is made available on our web portal. Outstanding performance obligations pertaining to orders received but for which the underlying report has not been issued are generally satisfied within a 30-day period.
We also generate test revenue through the sale of our precision oncology products, which is comprised primarily of sales of our distributed RUO and IVD products for therapy selection. We recognize revenue on these sales once shipment has occurred. Product sales are recorded net of discounts and other deductions. Billing terms are generally net 30 days.
Shipping and handling fees billed to customers are classified on the consolidated statements of operations and comprehensive loss in revenue. The associated shipping and handling costs are classified in cost of revenue.
Other revenue
Other revenue is primarily generated from pharma development services provided to biopharmaceutical companies related to companion diagnostic development as well as through collaboration agreements and genome network contracts.
Contracts for companion diagnostic development consist primarily of milestone-based payments along with annual fees and marked-up pass-through costs. The arrangements are treated as short-term contracts for revenue recognition purposes because they allow termination of the agreements by the customers with 30 to 120 days’ written notice without a termination penalty. Upon termination, customers are required to pay for the proportion of services provided under milestones that were in progress. We recognize revenue in an amount that reflects the consideration which we expect to receive in exchange for those goods or services. We recognize revenue over time based on the progress made toward achieving the performance obligation, utilizing both input or output methods, depending on the performance obligation, including labor hours expended, tests processed, or time elapsed, that measure our progress toward the achievement of the milestone.
We also enter into collaboration and genome network contracts. Collaboration agreements provide customers with diagnostic testing and related data aggregation reporting services that are provided over the contract term. Collaboration revenue is recognized as the data and reporting services are delivered to the customer. Genome network offerings consist of subscription services related to a proprietary software platform designed to connect patients, clinicians, advocacy organizations, researchers and therapeutic developers to accelerate the understanding, diagnosis and treatment of hereditary disease. Such services are recognized on a straight-line basis over the subscription periods. Amounts due under collaboration and genome network agreements are typically billable on net 30-day terms.
69
Business combinations
We apply Financial Accounting Standards Board, or FASB, Accounting Standards Codification, or ASC 805, Business Combinations, which requires recognition of assets acquired, liabilities assumed, and contingent consideration at their fair value on the acquisition date with subsequent changes recognized in earnings; requires acquisition-related expenses and restructuring costs to be recognized separately from the business combination and expensed as incurred; requires in-process research and development to be capitalized at fair value as an indefinite-lived intangible asset until completion or abandonment; and requires that changes in accounting for deferred tax asset valuation allowances and acquired income tax uncertainties after the measurement period be recognized as a component of provision for taxes.
We account for acquisitions of entities that include inputs and processes and have the ability to create outputs as business combinations. The tangible and identifiable intangible assets acquired and liabilities assumed in a business combination are recorded based on their estimated fair values as of the business combination date, including identifiable intangible assets which either arise from a contractual or legal right or are separable from goodwill. We base the estimated fair value of identifiable intangible assets acquired in a business combination on third-party valuations that use information and assumptions provided by our management, which consider our estimates of inputs and assumptions that a market participant would use. Any excess purchase price over the estimated fair value assigned to the net tangible and identifiable intangible assets acquired and liabilities assumed is recorded to goodwill. The use of alternative valuation assumptions, including estimated revenue projections, growth rates, estimated cost savings, cash flows, discount rates, estimated useful lives and probabilities surrounding the achievement of contingent milestones could result in different purchase price allocations and amortization expense in current and future periods.
In circumstances where an acquisition involves a contingent consideration arrangement that meets the definition of a liability under ASC Topic 480, Distinguishing Liabilities from Equity, we recognize a liability equal to the fair value of the contingent payments we expect to make as of the acquisition date. We remeasure this liability each reporting period and record changes in the fair value as a component of operating expenses.
Transaction costs associated with acquisitions are expensed as incurred in general and administrative expenses. Results of operations and cash flows of acquired companies are included in our operating results from the date of acquisition.
Goodwill
In accordance with ASC 350, Intangibles - Goodwill and Other, or ASC 350, we do not amortize goodwill or other intangible assets with indefinite lives but rather test them for impairment. ASC 350 requires us to perform an impairment review of our goodwill balance at least annually, which we do in the fourth quarter of each year for our single consolidated reporting unit, and whenever events or changes in circumstances indicate that the carrying amount of these assets may not be recoverable. We did not incur any goodwill impairment losses in any of the periods presented.
Stock-based compensation
We incur stock-based compensation expense for awards granted to employees and directors and for inducement awards granted in connection with our business acquisitions. Stock-based compensation expense is measured at the date of grant and is based on the estimated fair value of the award. Compensation cost is recognized as expense on a straight-line basis over the vesting period for options and restricted stock unit, or RSU, awards and on an accelerated basis for performance-based restricted stock unit, or PRSU, awards. We recognize stock-based compensation expense associated with PRSU grants when we determine the achievement of performance conditions is probable. In determining the fair value of stock options and Employee Stock Purchase Plan, or ESPP, purchases, we estimate the grant date fair value, and the resulting stock-based compensation expense, using the Black-Scholes option-pricing model. We estimate the grant date fair value of RSU and PRSU awards based on the grant date share price.
The Black-Scholes option-pricing model requires the use of highly subjective assumptions, which determine the fair value of stock-based awards. These assumptions include:
Expected term—The expected term represents the period that stock-based awards are expected to be outstanding. We use the simplified method to determine the expected term, which is based on the mid-point between the vesting date and the end of the contractual term.
70
Expected volatility—We estimate expected volatility based on the historical volatility of our common stock over a period equal to the expected term of awards and over the expected six-month term ESPP purchase periods.
Risk-free interest rate—The risk-free interest rate is based on the U.S. Treasury zero coupon issues in effect at the time of grant for periods corresponding with the expected term of an option.
Dividend yield—We have never paid dividends on our common stock and have no plans to pay dividends on our common stock. Therefore, we used an expected dividend yield of zero.
In addition to the Black-Scholes assumptions, we estimate our forfeiture rate based on an analysis of our actual forfeitures and will continue to evaluate the adequacy of the forfeiture rate based on actual forfeiture experience, analysis of employee turnover behavior and other factors. The impact from any forfeiture rate adjustment would be recognized in full in the period of adjustment and if the actual number of future forfeitures differs from our estimates, we might be required to record adjustments to stock-based compensation in future periods.
Income taxes
We use the asset and liability method of accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on the differences between the financial reporting and the tax bases of assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. A valuation allowance is provided when it is more likely than not that some portion or all of a deferred tax asset will not be realized. Significant judgment is required in determining the net valuation allowance which includes our evaluation of all available evidence including past operating results, estimates on future taxable income and acquisition-related tax assets and liabilities. As of December 31, 2020, we recorded a full valuation allowance on our net deferred tax assets because we expect that it is more likely than not that our deferred tax assets will not be realized in the foreseeable future. Should the actual amounts differ from our estimates, the amount of our valuation allowance could be materially impacted.
Results of operations
A discussion regarding our financial condition and results of operations for the year ended December 31, 2020 compared to the year ended December 31, 2019 is presented below. A discussion regarding our financial condition and results of operations for the year ended December 31, 2019 compared to the year ended December 31, 2018 can be found under Part II, Item 7 in our Annual Report on Form 10-K for the year ended December 31, 2019.
Comparison of the Years Ended December 31, 2020 and 2019
| Year Ended December 31, | Dollar Change | % Change | ||||||||||||||||||||||||
| 2020 | 2019 | |||||||||||||||||||||||||
| Revenue: | ||||||||||||||||||||||||||
| Test revenue | $ | 272,310 | $ | 212,473 | $ | 59,837 | 28% | |||||||||||||||||||
| Other revenue | 7,288 | 4,351 | 2,937 | 68% | ||||||||||||||||||||||
| Total revenue | 279,598 | 216,824 | 62,774 | 29% | ||||||||||||||||||||||
| Cost of revenue | 198,275 | 118,103 | 80,172 | 68% | ||||||||||||||||||||||
| Research and development | 240,605 | 141,526 | 99,079 | 70% | ||||||||||||||||||||||
| Selling and marketing | 168,317 | 122,237 | 46,080 | 38% | ||||||||||||||||||||||
| General and administrative | 324,573 | 79,070 | 245,503 | 310% | ||||||||||||||||||||||
| Loss from operations | (652,172) | (244,112) | (408,060) | 167% | ||||||||||||||||||||||
| Other expense, net | (32,332) | (3,891) | (28,441) | 731% | ||||||||||||||||||||||
| Interest expense | (29,766) | (12,412) | (17,354) | 140% | ||||||||||||||||||||||
| Net loss before taxes | (714,270) | (260,415) | (453,855) | 174% | ||||||||||||||||||||||
| Income tax benefit | (112,100) | (18,450) | (93,650) | 508% | ||||||||||||||||||||||
| Net loss | $ | (602,170) | $ | (241,965) | $ | (360,205) | 149% | |||||||||||||||||||
71
Revenue
The increase in revenue of $62.8 million for the year ended December 31, 2020 compared to the same period in 2019 was due primarily to increased billable volume from growth in our business as well as the contribution from businesses acquired in 2020, including ArcherDX in the fourth quarter of 2020. Billable units increased to approximately 659,000 during the year ended December 31, 2020 compared to 469,000 in the same period in 2019, an increase of 41%. Average revenue per unit decreased to $413 during the year ended December 31, 2020 compared to $453 in the same period in 2019, primarily due to changes in payer and product mix, the impact of the businesses acquired during 2020, particularly ArcherDX, as well as reductions in pricing for some payers as we focus on providing cost effective genetic testing.
Cost of revenue
The increase in the cost of revenue of $80.2 million for the year ended December 31, 2020 compared to the same period in 2019 was primarily due to costs associated with increased billable volume and the added costs related to businesses acquired in 2020, partially offset by the effect of cost efficiencies. For the year ended December 31, 2020, the number of units billed increased to approximately 659,000 from approximately 469,000 for the same period in 2019. Cost per billable unit was $299 in 2020 compared to $252 in 2019. The cost per unit increased primarily due to an increase in amortization of acquired intangible assets by $17.5 million as well as increased stock-based compensation by $4.2 million. The increase in the cost per unit was also due to changes in product mix, including the impact of the cost per unit of ArcherDX, as well as the influence of COVID-19. The increases were partially offset by production improvements that resulted in material efficiencies and automation and software improvements which reduced the medical interpretation time per report.
Research and development
The increase in research and development expense of $99.1 million for the year ended December 31, 2020 compared to the same period in 2019 was due to growth in the business and the effect of business acquisitions in 2020 and principally consisted of increases in personnel-related costs by $87.7 million, reflecting increased headcount as well as a $39.3 million increase in stock-based compensation; an increase in general lab expenses by $8.4 million; an increase in information technology costs by $4.8 million due to increased spending on networking equipment and software licenses; an increase by $4.7 million in professional fees; an increase by $2.4 million of depreciation and amortization; and an increase by $1.9 million in occupancy expenses. These cost increases were partially offset by a net increase of $9.6 million in allocations of resources from research and development to cost of revenue to support the increase in production volumes as well as a decrease in travel-related costs by $1.3 million due to a reduction in travel as a result of COVID-19.
Selling and marketing
The increase in selling and marketing expenses of $46.1 million for the year ended December 31, 2020 compared to the same period in 2019 was due to growth in the business and the effect of business acquisitions in 2020 and principally consisted of the following elements: an increase in personnel costs by $40.6 million due to increases in headcount; an increase by $3.7 million in allocations from other functional areas; an increase in marketing costs, principally for branding initiatives and advertising, by $2.0 million; an increase in information technology costs by $1.4 million; an increase in professional fees by $1.2 million; and an increase in depreciation and amortization by $1.2 million. These cost increases were partially offset by a decrease in travel expenses of $4.1 million due to a reduction in travel as a result of COVID-19.
General and administrative
The increase in general and administrative expenses of $245.5 million for the year ended December 31, 2020 compared to the same period in 2019 was primarily due to the growth of the business and the effect of business acquisitions in 2020 and principally consisted of the following elements: an increase in acquisition-related expense by $140.1 million, which includes $125.8 million of post-combination expense related to the acceleration of unvested equity from our acquisition of ArcherDX; an increase in fair value adjustments to contingent consideration by $54.4 million, primarily related to the development milestones for ArcherDX; an increase in personnel-related costs by $46.5 million primarily due to increases in headcount; an increase in legal and accounting costs by $5.2 million; an increase in information technology costs by $2.7 million due primarily to computer equipment and software purchases to support headcount growth; and an increase in depreciation and amortization by $0.9 million.
These cost increases were offset by increased allocations of technology and facilities-related expenses to other functional areas of $7.1 million.
72
Other expense, net
The increase in other expense, net of $28.4 million for the year ended December 31, 2020 compared to the same period in 2019 was principally due to fair value adjustments related to our stock payable liabilities of $37.5 million due to the increase in the price of our common stock partially offset by a reduction of debt extinguishment costs of $8.9 million incurred in September 2019 with no similar expense in 2020, $3.8 million received under the CARES Act during 2020, and decreases in interest income from our cash equivalents and marketable securities.
Interest expense
The increase in interest expense of $17.4 million for the year ended December 31, 2020 compared to the same period in 2019 was due principally to increased borrowings under our debt facilities as compared to the prior year period. See Note 8, “Commitments and contingencies” in Notes to Consolidated Financial Statements in Part II, Item 8 of this Annual Report.
Income tax benefit
The increase in income tax benefit of $93.7 million for the year ended December 31, 2020 compared to the same period in 2019 was due to net deferred tax liabilities assumed in connection with our acquisitions of YouScript Incorporated and ArcherDX which provided a future source of income to support the realization of our deferred tax assets and resulted in a partial release of our valuation allowance. As the short period tax returns for our 2020 acquisitions have not yet been filed, material changes to the tax returns may have a material impact on the net deferred tax liabilities assumed in connection with the acquisitions and the related income tax benefit.
Liquidity and capital resources
Liquidity and capital expenditures
We have incurred net losses since our inception. For the years ended December 31, 2020, 2019 and 2018, our net losses were $602.2 million, $242.0 million and $129.4 million, respectively, and we expect to incur additional losses in the future. At December 31, 2020, we had an accumulated deficit of $1.4 billion. While our revenue has increased over time, we may never achieve revenue sufficient to offset our expenses.
Since inception, our operations have been financed primarily by fees collected from our customers, net proceeds from sales of our capital stock as well as borrowing from debt facilities and the issuance of Convertible Senior Notes.
In March 2019, we issued, in an underwritten public offering, an aggregate of 10.4 million shares of our common stock at a price of $19.00 per share, for gross proceeds of $196.7 million and net proceeds of $184.5 million. During 2019, we issued 0.8 million shares of common stock at an average price of $25.71 per share in "at the market" offerings for aggregate proceeds of $20.2 million and net proceeds of $19.5 million. In April 2020, we issued, in an underwritten public offering, an aggregate of 20.4 million shares of our common stock at a price of $9.00 per share, for gross proceeds of $184.0 million and net proceeds of approximately $173.0 million. In 2020, we issued approximately 3.6 million shares of common stock at an average price of $26.33 per share in an "at the market" offering for aggregate proceeds of $93.7 million and net proceeds of $90.7 million. In January 2021, we issued, in an underwritten public offering, an aggregate of 8.9 million shares of our common stock at a price of $51.50 per share, for gross proceeds of $460.0 million and net proceeds of approximately $434.3 million.
In September 2019, we issued $350.0 million of aggregate principal amount of Convertible Senior Notes, which bear cash interest at a rate of 2.0% per year. Also in September 2019, we used the funds received through the issuance of our Convertible Senior Notes to settle our Note Purchase Agreement we entered into in November 2018.
In October 2020 in connection with our acquisition of ArcherDX, we issued $275.0 million of our common stock in a private placement at a price of $16.85 per share to a syndicate of life sciences investors. We also entered into a credit facility to borrow $135.0 million. The private placement and credit facility closed concurrently with the merger in October 2020. In connection with the credit facility, we issued warrants to purchase 1.0 million shares of our common stock at an exercise price of $16.85 per share which were exercised in October 2020 on a net exercise basis. The terms of this credit facility restrict our ability to incur certain indebtedness, pay dividends, make acquisitions and take other actions.
At December 31, 2020 and 2019, we had $360.7 million and $398.0 million, respectively, of cash, cash equivalents, restricted cash and marketable securities.
73
Our primary uses of cash are to fund our operations as we continue to grow our business, enter into partnerships and acquire businesses and technologies. Cash used to fund operating expenses is affected by the timing of when we pay expenses, as reflected in the change in our outstanding accounts payable and accrued expenses. We estimate our capital expenditures will be approximately $30.0 million for 2021.
We have incurred substantial losses since inception, and we expect to continue to incur losses in the future. We believe our existing cash, cash equivalents and marketable securities as of December 31, 2020 and fees collected from the sale of our tests will be sufficient to meet our anticipated cash requirements for the foreseeable future.
We may need additional funding to finance operations prior to achieving profitability or should we make additional acquisitions. We regularly consider fundraising opportunities and will determine the timing, nature and size of future financings based upon various factors, including market conditions and our operating plans. We may in the future elect to finance operations by selling equity or debt securities or borrowing money. We also may elect to finance future acquisitions. If we issue equity securities, dilution to stockholders may result. Any equity securities issued may also provide for rights, preferences or privileges senior to those of holders of our common stock. If we raise funds by issuing additional debt securities, these debt securities would have rights, preferences and privileges senior to those of holders of our common stock. In addition, the terms of additional debt securities or borrowings could impose significant restrictions on our operations. If additional funding is required, there can be no assurance that additional funds will be available to us on acceptable terms on a timely basis, if at all. If we are unable to obtain additional funding when needed, we may need to curtail planned activities to reduce costs. Doing so will likely have an unfavorable effect on our ability to execute on our business plan and have an adverse effect on our business, results of operations and future prospects.
The following table summarizes our cash flows (in thousands):
| Year Ended December 31, | ||||||||||||||||||||
| 2020 | 2019 | 2018 | ||||||||||||||||||
| Cash used in operating activities | $ | (298,502) | $ | (145,053) | $ | (92,220) | ||||||||||||||
| Cash provided by (used in) investing activities | (400,583) | (280,310) | 35,773 | |||||||||||||||||
| Cash provided by financing activities | 672,993 | 464,771 | 157,152 | |||||||||||||||||
| Net increase (decrease) in cash, cash equivalents and restricted cash | $ | (26,092) | $ | 39,408 | $ | 100,705 | ||||||||||||||
Cash flows from operating activities
For the year ended December 31, 2020, cash used in operating activities was $298.5 million and principally resulted from our net loss of $602.2 million and $112.1 million related to our income tax benefit generated from business combinations completed in 2020 partially offset by non-cash charges of $158.7 million for stock-based compensation, $92.3 million in remeasurements of liabilities associated with business combinations such as contingent consideration, $91.0 million related to post-combination expense due to the acceleration of unvested equity in the acquisition of ArcherDX, $39.1 million for depreciation and amortization, $17.2 million of amortization of debt discount and issuance costs and $1.4 million of other adjustments. The net effect on cash for changes in net operating assets was an inflow of cash of $16.0 million due principally to increases in accounts payable and accrued liabilities partially offset by increases in inventory and accounts receivable due to timing of collections.
For the year ended December 31, 2019, cash used in operating activities of $145.1 million principally resulted from our net loss of $242.0 million and $18.5 million related to our income tax benefit generated from business combinations completed in 2019 offset by non-cash charges of $75.9 million for stock-based compensation, $16.2 million for depreciation and amortization, $8.9 million for debt extinguishment costs related to the settlement of our 2018 Note Purchase Agreement and $1.1 million of other adjustments. The net effect on cash for changes in net operating assets was a use of cash of $8.8 million due principally to increases in accrued liabilities which include acquisition-related liabilities for 2019 business acquisitions partially offset by increases in accounts receivable due to timing of collections and increases in prepaid expenses and other current assets.
74
For the year ended December 31, 2018, cash used in operating activities of $92.2 million principally resulted from our net loss of $129.4 million offset by non-cash charges of $20.9 million for stock-based compensation, $13.5 million for depreciation and amortization, $5.3 million related to debt extinguishment costs, $2.9 million of impairment losses related to a collaboration agreement, $0.8 million of other non-cash adjustments and $0.4 million for remeasurements of liabilities associated with business combinations, all partially offset by a $2.9 million benefit from income taxes resulting from the completion of our analysis of historical net operating losses for CombiMatrix Corporation. The net effect on cash of changes in net operating assets was a use of cash of $3.8 million due principally to the effect of increase in accounts receivable due to timing of collections partially offset by an increase in accrued and other liabilities.
Cash flows from investing activities
For the year ended December 31, 2020, cash used in investing activities of $400.6 million was primarily related to net cash used to acquire Orbicule BV ("Diploid"), Genelex, YouScript and ArcherDX of $383.8 million, purchases of property and equipment of $22.9 million, and other cash outflows of $4.0 million, all partially offset by net sales and maturities of marketable securities of $10.1 million.
For the year ended December 31, 2019, cash used in investing activities of $280.3 million resulted primarily from purchases of marketable securities exceeding proceeds from maturities and sales of marketable securities by $226.4 million, net cash used to acquire Singular Bio, Jungla Inc., and Clear Genetics, Inc. of $33.8 million and purchases of property and equipment of $20.0 million.
For the year ended December 31, 2018, cash provided by investing activities of $35.8 million resulted primarily from proceeds from maturities and sales of marketable securities exceeding purchases of marketable securities by $42.7 million and purchases of property and equipment of $6.0 million.
Cash flows from financing activities
For the year ended December 31, 2020, cash provided by financing activities of $673.0 million consisted of cash received from issuances of common stock totaling $284.2 million, including cash received from shares issued through a private placement in October 2020 upon the close of the ArcherDX acquisition, exercises of stock options and employee stock plan purchases; net proceeds from the public offerings of common stock of $263.7 million; and net proceeds from debt financings of $129.2 million. These cash inflows were partially offset by other cash outflows of $4.1 million.
For the year ended December 31, 2019, cash provided by financing activities of $464.8 million consisted of net proceeds from the issuance of Convertible Senior Notes of $339.9 million, net proceeds from the public offerings of common stock of $204.0 million and cash received from issuances of common stock totaling $9.5 million, including cash received from exercises of stock options of $3.5 million and employee stock plan purchases of $5.8 million. These cash inflows were partially offset by payments related to the settlement of our Note Purchase Agreement through repayment of loan obligations of $75.0 million and payment of debt extinguishment costs of $10.6 million, as well as finance lease payments of $2.1 million.
For the year ended December 31, 2018, cash provided by financing activities of $157.2 million consisted of net proceeds from the public offerings of common stock of $112.4 million, net proceeds of $93.9 million from the second term loan under the Amended 2017 Loan Agreement and from the 2018 Note Purchase Agreement, and cash received from issuances of common stock totaling $17.5 million (which includes $6.5 million received from exercises of warrants issued pursuant to the acquisition of CombiMatrix, $5.0 million received pursuant to the Securities Purchase Agreement entered into in connection with our 2018 Note Purchase Agreement, employee stock purchases of $3.2 million, and stock option exercises of $2.7 million). These cash inflows were partially offset by loan payments of $60.0 million to extinguish our 2017 Loan Agreement, payments of $4.6 million related to the extinguishment of our 2017 Loan Agreement and related amendments and capital lease payments of $2.1 million.
75
Contractual obligations
The following table summarizes our contractual obligations, including interest, as of December 31, 2020 (in thousands):
| Contractual obligations: | 2021 | 2022 and 2023 | 2024 and 2025 | 2026 and beyond | Total | |||||||||||||||||||||||||||
| Operating leases | $ | 14,338 | $ | 27,017 | $ | 25,079 | $ | 9,499 | $ | 75,933 | ||||||||||||||||||||||
| Finance leases | 2,006 | 3,107 | 26 | — | 5,139 | |||||||||||||||||||||||||||
| Convertible Senior Notes | — | — | 350,000 | — | 350,000 | |||||||||||||||||||||||||||
| 2020 Term Loan | — | — | 135,000 | — | 135,000 | |||||||||||||||||||||||||||
| Purchase commitments | 23,064 | 39,823 | 13,750 | 25,501 | 102,138 | |||||||||||||||||||||||||||
| Total | $ | 39,408 | $ | 69,947 | $ | 523,855 | $ | 35,000 | $ | 668,210 | ||||||||||||||||||||||
See Note 8, “Commitments and contingencies” in Notes to Consolidated Financial Statements in Part II, Item 8 of this Annual Report for additional details regarding our leases, Convertible Senior Notes, 2020 Term Loan and purchase commitments.
Off-balance sheet arrangements
We have not entered into any off-balance sheet arrangements.
Recent accounting pronouncements
See “Recent accounting pronouncements” in Note 2, “Summary of significant accounting policies” in the Notes to Consolidated Financial Statements for a discussion of recently adopted accounting pronouncements and accounting pronouncements not yet adopted, and their expected effect on our financial position and results of operations.
76
ITEM 7A. Quantitative and Qualitative Disclosures about Market Risk.
We are exposed to market risks in the ordinary course of our business. These risks primarily relate to interest rates. Our cash, cash equivalents, restricted cash and marketable securities totaled $360.7 million at December 31, 2020, and consisted primarily of money market funds, U.S. treasury notes, and U.S. government agency securities. Such interest-bearing instruments carry a degree of risk; however, because our investments are primarily high-quality credit instruments with short-term durations with high-quality institutions, we have not been exposed to, nor do we anticipate being exposed to, material risks due to changes in interest rates. At December 31, 2020, a hypothetical 1.0% (100 basis points) increase or decrease in interest rates would not have resulted in a material change in the fair value of our cash equivalents and portfolio of marketable securities. Fluctuations in the value of our cash equivalents and portfolio of marketable securities caused by a change in interest rates (gains or losses on the carrying value) are recorded in other comprehensive gain (loss) and are realized only if we sell the underlying securities prior to maturity or declines in fair value are determined to be other-than-temporary.
Our 2020 Term Loan bears interest at an annual rate equal to LIBOR, subject to a 2.00% LIBOR floor, plus a margin of 8.75% and is therefore sensitive to changes in interest rates. We currently do not use interest rate derivative instruments to manage our exposure to interest rate fluctuations.
Although our Convertible Senior Notes are based on a fixed rate, changes in interest rates could impact their fair market value. As of December 31, 2020, the fair market value of the Convertible Senior Notes was $586.0 million. For additional information about the Convertible Senior Notes, see Note 8, “Commitments and contingencies” in Notes to Consolidated Financial Statements in Part II, Item 8 of this Annual Report.
77
ITEM 8. Consolidated Financial Statements and Supplementary Data.
Index to Consolidated Financial Statements
| Page | |||||
78
Report of Independent Registered Public Accounting Firm
To the Stockholders and Board of Directors of Invitae Corporation
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Invitae Corporation (the Company) as of December 31, 2020 and 2019, the related consolidated statements of operations, comprehensive loss, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2020, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2020 and 2019, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2020, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company's internal control over financial reporting as of December 31, 2020, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated February 26, 2021 expressed an unqualified opinion thereon.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
79
| Measurement of test revenue | |||||
| Description of the Matter | During the year ended December 31, 2020, the Company’s test revenue subject to estimation was $181.0 million. As discussed in Note 3 of the consolidated financial statements, test revenue is recognized when the performance obligation is complete, generally upon delivery of the underlying clinical report or when the report is made available to the customer on the Company’s website. The amounts recognized are based on estimates of the consideration that the Company expects to receive, and such estimates are adjusted and subsequently recorded until fully settled. Auditing the measurement of the Company’s test revenue was complex and judgmental due to the significant estimation required in determining the amount expected to be collected for each test. In particular, the estimate of revenue for tests billed to insurance carriers is affected by assumptions in payer behavior such as changes in historical payment patterns, contract provisions and government and private insurance reimbursement policies. | ||||
| How We Addressed the Matter in Our Audit | We obtained an understanding, evaluated the design and tested the operating effectiveness of controls over the Company’s revenue recognition process. As part of our testing, we considered controls over management’s review of the significant assumptions and inputs used in the determination of the amount expected to be collected. We also tested controls used by management to compare the current and historical data used in making the estimates for completeness and accuracy. Our audit procedures over the Company’s test revenue included, among others, assessing valuation methodologies and models and testing the significant assumptions above and the underlying data used by the Company in its analysis. We agreed a sample of transactions to the payer contract terms. We compared the significant assumptions above and inputs used by management to changes in the Company’s contracted rates, government and private insurance payer collection trends, and other relevant factors. We assessed the historical accuracy of the cash collections used in the Company’s revenue models and assessed the completeness of adjustments to estimates of future cash collections as a result of significant contract amendments and changes in collection trends. | ||||
| Valuation of intangible assets associated with business acquisitions | |||||
| Description of the Matter | As described in Note 4 to the consolidated financial statements, the Company completed several business acquisitions during 2020. As a result of the acquisitions, the Company recorded goodwill of $1,736.8 million, and intangible assets of $880.4 million. The acquisitions were accounted for as business combinations. Auditing the Company’s accounting for the acquisitions was challenging as the determination of the fair value of the intangible assets acquired required management to make subjective estimates and assumptions. The Company used an income approach to measure the acquired intangible assets. The valuation of the intangible assets is subject to higher estimation uncertainty due to management’s judgments in determining significant assumptions that included assumed revenue growth, estimated cost savings and discount rates. Changes in these significant assumptions could have a significant effect on the fair value of the intangible assets. | ||||
| How We Addressed the Matter in Our Audit | We tested the design and operating effectiveness of internal controls over the Company’s process for accounting for acquisitions. For example, we tested controls over management’s review of the valuation of intangible assets, including the review of the valuation model and significant assumptions used in the valuation. Our audit procedures related to the valuation of intangible assets included, among others, utilizing a valuation specialist to assist in evaluating the appropriateness of the Company’s valuation models and evaluating the reasonableness of significant assumptions used such as the revenue growth, estimated cost savings and the discount rates as compared to industry and market data and historical results. We also evaluated whether the assumptions used were reasonable by comparing them to the past performance of prior acquisitions, current industry data and current market forecasts, and whether such assumptions were consistent with evidence obtained in other areas of the audit. | ||||
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 2013.
Redwood City, California
February 26, 2021
80
INVITAE CORPORATION
Consolidated Balance Sheets
(in thousands, except par value data)
| December 31, | |||||||||||
| 2020 | 2019 | ||||||||||
| Assets | |||||||||||
| Current assets: | |||||||||||
| Cash and cash equivalents | $ | 124,794 | $ | 151,389 | |||||||
| Marketable securities | 229,186 | 240,436 | |||||||||
| Accounts receivable | 47,722 | 32,541 | |||||||||
| Inventory | 32,030 | 6,648 | |||||||||
| Prepaid expenses and other current assets | 20,200 | 11,384 | |||||||||
| Total current assets | 453,932 | 442,398 | |||||||||
| Property and equipment, net | 66,102 | 37,747 | |||||||||
| Operating lease assets | 45,109 | 36,640 | |||||||||
| Restricted cash | 6,686 | 6,183 | |||||||||
| Intangible assets, net | 981,845 | 125,175 | |||||||||
| Goodwill | 1,863,623 | 126,777 | |||||||||
| Other assets | 13,188 | 6,681 | |||||||||
| Total assets | $ | 3,430,485 | $ | 781,601 | |||||||
| Liabilities and stockholders’ equity | |||||||||||
| Current liabilities: | |||||||||||
| Accounts payable | $ | 25,203 | $ | 10,321 | |||||||
| Accrued liabilities | 86,058 | 64,814 | |||||||||
| Operating lease obligation | 8,789 | 4,870 | |||||||||
| Finance lease obligation | 1,695 | 1,855 | |||||||||
| Total current liabilities | 121,745 | 81,860 | |||||||||
| Operating lease obligation, net of current portion | 48,357 | 42,191 | |||||||||
| Finance lease obligation, net of current portion | 3,123 | 1,155 | |||||||||
| Debt | 104,449 | 0 | |||||||||
| Convertible senior notes, net | 283,724 | 268,755 | |||||||||
| Deferred tax liability | 51,538 | 0 | |||||||||
| Other long-term liabilities | 841,256 | 8,000 | |||||||||
| Total liabilities | 1,454,192 | 401,961 | |||||||||
| Commitments and contingencies (Note 8) | 0 | 0 | |||||||||
| Stockholders’ equity: | |||||||||||
| Preferred stock, $0.0001 par value: 20,000 shares authorized; 125 shares issued and outstanding as of December 31, 2020 and 2019 | 0 | 0 | |||||||||
| Common stock, $0.0001 par value: 400,000 shares authorized; 185,886 and 98,796 shares issued and outstanding as of December 31, 2020 and 2019, respectively | 19 | 10 | |||||||||
| Accumulated other comprehensive income (loss) | 1 | (9) | |||||||||
| Additional paid-in capital | 3,337,120 | 1,138,316 | |||||||||
| Accumulated deficit | (1,360,847) | (758,677) | |||||||||
| Total stockholders’ equity | 1,976,293 | 379,640 | |||||||||
| Total liabilities and stockholders’ equity | $ | 3,430,485 | $ | 781,601 | |||||||
The accompanying notes are an integral part of these financial statements.
81
INVITAE CORPORATION
Consolidated Statements of Operations
(in thousands, except per share data)
| Year Ended December 31, | |||||||||||||||||
| 2020 | 2019 | 2018 | |||||||||||||||
| Revenue: | |||||||||||||||||
| Test revenue | $ | 272,310 | $ | 212,473 | $ | 144,560 | |||||||||||
| Other revenue | 7,288 | 4,351 | 3,139 | ||||||||||||||
| Total revenue | 279,598 | 216,824 | 147,699 | ||||||||||||||
| Cost of revenue | 198,275 | 118,103 | 80,105 | ||||||||||||||
| Research and development | 240,605 | 141,526 | 63,496 | ||||||||||||||
| Selling and marketing | 168,317 | 122,237 | 74,428 | ||||||||||||||
| General and administrative | 324,573 | 79,070 | 52,227 | ||||||||||||||
| Loss from operations | (652,172) | (244,112) | (122,557) | ||||||||||||||
| Other expense, net | (32,332) | (3,891) | (2,568) | ||||||||||||||
| Interest expense | (29,766) | (12,412) | (7,030) | ||||||||||||||
| Net loss before taxes | (714,270) | (260,415) | (132,155) | ||||||||||||||
| Income tax benefit | (112,100) | (18,450) | (2,800) | ||||||||||||||
| Net loss | $ | (602,170) | $ | (241,965) | $ | (129,355) | |||||||||||
| Net loss per share, basic and diluted | $ | (4.47) | $ | (2.66) | $ | (1.94) | |||||||||||
| Shares used in computing net loss per share, basic and diluted | 134,587 | 90,859 | 66,747 | ||||||||||||||
The accompanying notes are an integral part of these financial statements.
82
INVITAE CORPORATION
Consolidated Statements of Comprehensive Loss
(in thousands)
| Year Ended December 31, | |||||||||||||||||
| 2020 | 2019 | 2018 | |||||||||||||||
| Net loss | $ | (602,170) | $ | (241,965) | $ | (129,355) | |||||||||||
| Other comprehensive income (loss): | |||||||||||||||||
| Unrealized income (loss) on available-for-sale marketable securities, net of tax | 10 | (4) | 166 | ||||||||||||||
| Comprehensive loss | $ | (602,160) | $ | (241,969) | $ | (129,189) | |||||||||||
The accompanying notes are an integral part of these financial statements.
83
INVITAE CORPORATION
Consolidated Statements of Stockholders’ Equity
(in thousands)
| Year Ended December 31, | |||||||||||||||||
| 2020 | 2019 | 2018 | |||||||||||||||
| Common stock: | |||||||||||||||||
| Balance, beginning of period | $ | 10 | $ | 8 | $ | 5 | |||||||||||
| Common stock issued | 9 | 2 | 3 | ||||||||||||||
| Balance, end of period | 19 | 10 | 8 | ||||||||||||||
| Accumulated other comprehensive income (loss): | |||||||||||||||||
| Balance, beginning of period | (9) | (5) | (171) | ||||||||||||||
| Unrealized income (loss) on available-for-sale marketable securities, net of tax | 10 | (4) | 166 | ||||||||||||||
| Balance, end of period | 1 | (9) | (5) | ||||||||||||||
| Additional paid-in capital: | |||||||||||||||||
| Balance, beginning of period | 1,138,316 | 678,548 | 520,558 | ||||||||||||||
| Common stock issued in private placement, net | 263,628 | — | 5,353 | ||||||||||||||
| Common stock issued in connection with public offering, net | 263,685 | 204,024 | 112,438 | ||||||||||||||
| Common stock issued on exercise of stock options, net | 10,730 | 3,456 | 2,741 | ||||||||||||||
| Common stock issued pursuant to exercises of warrants | 974 | 181 | 6,539 | ||||||||||||||
| Common stock issued pursuant to employee stock purchase plan | 8,871 | 5,833 | 3,231 | ||||||||||||||
| Common stock issued or issuable pursuant to acquisitions | 1,524,227 | 133,942 | 6,455 | ||||||||||||||
| Equity component of convertible senior notes, net | — | 75,488 | — | ||||||||||||||
| Warrants issued pursuant to loan agreement | 27,000 | — | 383 | ||||||||||||||
| Stock-based compensation expense | 110,076 | 36,844 | 20,850 | ||||||||||||||
| Reclassification of stock payable liabilities | (10,387) | — | — | ||||||||||||||
| Balance, end of period | 3,337,120 | 1,138,316 | 678,548 | ||||||||||||||
| Accumulated deficit: | |||||||||||||||||
| Balance, beginning of period | (758,677) | (516,712) | (398,598) | ||||||||||||||
| Cumulative effect of accounting change | — | — | 11,241 | ||||||||||||||
| Net loss | (602,170) | (241,965) | (129,355) | ||||||||||||||
| Balance, end of period | (1,360,847) | (758,677) | (516,712) | ||||||||||||||
| Total stockholders' equity | $ | 1,976,293 | $ | 379,640 | $ | 161,839 | |||||||||||
The accompanying notes are an integral part of these financial statements.
84
INVITAE CORPORATION
Consolidated Statements of Cash Flows
(in thousands)
| Year Ended December 31, | |||||||||||||||||
| 2020 | 2019 | 2018 | |||||||||||||||
| Cash flows from operating activities: | |||||||||||||||||
| Net loss | $ | (602,170) | $ | (241,965) | $ | (129,355) | |||||||||||
| Adjustments to reconcile net loss to net cash used in operating activities: | |||||||||||||||||
| Depreciation and amortization | 39,050 | 16,206 | 13,540 | ||||||||||||||
| Stock-based compensation | 158,747 | 75,948 | 20,850 | ||||||||||||||
| Amortization of debt discount and issuance costs | 17,204 | 4,416 | 0 | ||||||||||||||
| Impairment losses | 0 | 0 | 2,925 | ||||||||||||||
| Remeasurements of liabilities associated with business combinations | 92,348 | 0 | 0 | ||||||||||||||
| Benefit from income taxes | (112,100) | (18,450) | (2,862) | ||||||||||||||
| Debt extinguishment costs | 0 | 8,926 | 5,266 | ||||||||||||||
| Post-combination expense for acceleration of unvested equity | 91,021 | 0 | 0 | ||||||||||||||
| Other | 1,425 | 1,095 | 1,168 | ||||||||||||||
| Changes in operating assets and liabilities, net of businesses acquired: | |||||||||||||||||
| Accounts receivable | (2,814) | (6,131) | (5,291) | ||||||||||||||
| Inventory | (7,832) | 1,645 | (2,848) | ||||||||||||||
| Prepaid expenses and other current assets | (2,010) | (6,624) | 1,403 | ||||||||||||||
| Other assets | 895 | 2,026 | (163) | ||||||||||||||
| Accounts payable | 10,186 | 1,558 | (417) | ||||||||||||||
| Accrued expenses and other long-term liabilities | 17,548 | 16,297 | 3,564 | ||||||||||||||
| Net cash used in operating activities | (298,502) | (145,053) | (92,220) | ||||||||||||||
| Cash flows from investing activities: | |||||||||||||||||
| Purchases of marketable securities | (280,258) | (260,917) | (9,680) | ||||||||||||||
| Proceeds from sales of marketable securities | 12,832 | 0 | 19,965 | ||||||||||||||
| Proceeds from maturities of marketable securities | 277,487 | 34,500 | 32,458 | ||||||||||||||
| Acquisition of businesses, net of cash acquired | (383,753) | (33,846) | 0 | ||||||||||||||
| Purchases of property and equipment | (22,865) | (20,047) | (5,970) | ||||||||||||||
| Other | (4,026) | 0 | (1,000) | ||||||||||||||
| Net cash provided by (used in) investing activities | (400,583) | (280,310) | 35,773 | ||||||||||||||
| Cash flows from financing activities: | |||||||||||||||||
| Proceeds from public offerings of common stock, net of issuance costs | 263,688 | 204,024 | 112,441 | ||||||||||||||
| Proceeds from issuance of common stock, net | 284,203 | 9,470 | 17,511 | ||||||||||||||
| Proceeds from issuance of convertible senior notes, net | 0 | 339,900 | 0 | ||||||||||||||
| Proceeds from issuance of debt, net | 129,214 | 0 | 93,909 | ||||||||||||||
| Payments of debt extinguishment costs | 0 | (10,638) | (4,609) | ||||||||||||||
| Loan payments | 0 | (75,000) | (60,000) | ||||||||||||||
| Other | (4,112) | (2,985) | (2,100) | ||||||||||||||
| Net cash provided by financing activities | 672,993 | 464,771 | 157,152 | ||||||||||||||
| Net increase (decrease) in cash, cash equivalents and restricted cash | (26,092) | 39,408 | 100,705 | ||||||||||||||
| Cash, cash equivalents and restricted cash at beginning of period | 157,572 | 118,164 | 17,459 | ||||||||||||||
| Cash, cash equivalents and restricted cash at end of period | $ | 131,480 | $ | 157,572 | $ | 118,164 | |||||||||||
| Supplemental cash flow information: | |||||||||||||||||
| Interest paid | $ | 12,130 | $ | 4,731 | $ | 6,231 | |||||||||||
| Supplemental cash flow information of non-cash investing and financing activities: | |||||||||||||||||
| Equipment acquired through finance leases | $ | 4,463 | $ | 1,892 | $ | 0 | |||||||||||
| Purchases of property and equipment in accounts payable and accrued liabilities | $ | 1,869 | $ | 2,422 | $ | 510 | |||||||||||
| Warrants issued pursuant to debt agreement | $ | 27,000 | $ | 0 | $ | 383 | |||||||||||
| Common stock issued for acquisitions | $ | 1,157,958 | $ | 108,573 | $ | 6,445 | |||||||||||
| Consideration payable for acquisitions | $ | 940,829 | $ | 21,449 | $ | 0 | |||||||||||
| Operating lease assets obtained in exchange for lease obligations, net | $ | 14,058 | $ | 4,261 | $ | — | |||||||||||
The accompanying notes are an integral part of these financial statements.
85
INVITAE CORPORATION
Notes to Consolidated Financial Statements
1. Organization and description of business
Invitae Corporation ("Invitae," “the Company," "we," "us," and "our") was incorporated in the State of Delaware on January 13, 2010, as Locus Development, Inc. and we changed our name to Invitae Corporation in 2012. We offer high-quality, comprehensive, affordable genetic testing across multiple clinical areas, including hereditary cancer, cardiology, neurology, pediatrics, personalized oncology, metabolic conditions and rare diseases. To augment our offering and realize our mission, we have acquired multiple assets including 4 businesses in 2020, which expanded our suite of genome management offerings and established a broader entry into oncology therapy selection and personalized cancer monitoring.
In October 2020, we completed the acquisition of ArcherDX, Inc. (“ArcherDX”). ArcherDX is a genomics company democratizing precision oncology by offering a suite of products and services that are accurate, personal, actionable and easy to use in local settings, thereby empowering clinicians to control the sample, data, patient care and economics. ArcherDX’s development platform, including its proprietary Anchored Multiplex PCR, ("AMP"), chemistry at the core, is enabling clinical tests and services that allow for therapy selection and cancer monitoring in community locations for the first time at scale. Invitae operates in 1 segment.
2. Summary of significant accounting policies
Principles of consolidation
Our consolidated financial statements include our accounts and the accounts of our wholly-owned subsidiaries. All intercompany balances and transactions have been eliminated in consolidation.
Use of estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make judgments, estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent liabilities as of the date of the financial statements and the reported amounts of revenue and expenses during the reporting period. We base these estimates on current facts, historical and anticipated results, trends and various other assumptions that we believe are reasonable under the circumstances, including assumptions as to future events. Actual results could differ materially from those judgments, estimates and assumptions. We evaluate our estimates on an ongoing basis.
Significant estimates and assumptions made by management include the determination of:
•revenue recognition;
•the fair value of assets and liabilities associated with business combinations;
•the impairment assessment of goodwill and intangible assets;
•the valuation of our 2.00% convertible senior notes due 2024 issued in September 2019 ("Convertible Senior Notes");
•the recoverability of long-lived assets;
•our incremental borrowing rates used to calculate our lease balances;
•stock-based compensation expense and the fair value of awards and warrants issued; and
•income tax uncertainties.
86
Concentrations of credit risk and other risks and uncertainties
Financial instruments that potentially subject us to a concentration of credit risk consist of cash, cash equivalents, marketable securities and accounts receivable. Our cash and cash equivalents are held by financial institutions in the United States. Such deposits may exceed federally insured limits.
Significant customers are those that represent 10% or more of our total revenue for each year presented on the consolidated statements of operations. Our revenue from significant customers as a percentage of our total revenue was as follows:
| Year Ended December 31, | ||||||||||||||||||||
| 2020 | 2019 | 2018 | ||||||||||||||||||
| Medicare | 19 | % | 25 | % | 22 | % | ||||||||||||||
No customers represented more than 10% of accounts receivable as of December 31, 2020 or 2019.
Cash, cash equivalents, and restricted cash
We consider all highly liquid investments with original maturities of three months or less from the date of purchase to be cash equivalents. Cash equivalents consist primarily of amounts invested in money market funds, U.S. treasury notes and government agency securities.
Restricted cash consists primarily of money market funds that secure irrevocable standby letters of credit that serve as collateral for security deposits for our facility leases.
The following table provides a reconciliation of cash, cash equivalents and restricted cash reported within the consolidated balance sheets that sum to the total of the same amounts shown in the consolidated statements of cash flows (in thousands):
| December 31, | |||||||||||
| 2020 | 2019 | ||||||||||
| Cash and cash equivalents | $ | 124,794 | $ | 151,389 | |||||||
| Restricted cash | 6,686 | 6,183 | |||||||||
| Total cash, cash equivalents and restricted cash | $ | 131,480 | $ | 157,572 | |||||||
Marketable securities
All marketable securities have been classified as “available-for-sale” and are carried at estimated fair value as determined based upon quoted market prices or pricing models for similar securities. Management determines the appropriate classification of its marketable debt securities at the time of purchase and reevaluates such designation at each balance sheet date. Short-term marketable securities have maturities one year or less at the balance sheet date. Unrealized gains and losses are excluded from earnings and are reported as a component of other comprehensive loss. Realized gains and losses and impairments, if any, on available-for-sale securities are included in other expense, net. The cost of securities sold is based on the specific-identification method. Interest on marketable securities is included in other income (expense), net.
For marketable securities in an unrealized loss position, we assess our intent to sell, or whether it is more likely than not that we will be required to sell the security before recovery of its amortized cost basis. If either of these criteria are met, the security’s amortized cost basis is written down to fair value through other income (expense), net.
Accounts receivable
We receive payment from patients, biopharmaceutical partners, third-party payers and other business-to-business customers. See Note 3, "Revenue, accounts receivable and deferred revenue" for further information.
Deferred revenue
We record a contract liability when cash payments are received or due in advance of our performance related to one or more performance obligations. See Note 3, "Revenue, accounts receivable and deferred revenue" for further information.
87
Inventory
Our inventory consists of raw materials, work in progress, and finished goods, which are stated at the lower of cost or net realizable value on a first-in, first-out basis. We periodically analyze our inventory levels and expiration dates, and write down inventory that has become obsolete, inventory that has a cost basis in excess of its net realizable value, and inventory in excess of expected sales requirements as cost of revenue. We record an allowance for obsolete inventory using an estimate based on historical trends and evaluation of near-term expirations.
Business combinations
We apply Financial Accounting Standards Board ("FASB"), Accounting Standards Codification ("ASC") 805, Business Combinations, which requires recognition of assets acquired, liabilities assumed, and contingent consideration at their fair value on the acquisition date with subsequent changes recognized in earnings; requires acquisition-related expenses and restructuring costs to be recognized separately from the business combination and expensed as incurred; requires in-process research and development to be capitalized at fair value as an indefinite-lived intangible asset until completion or abandonment; and requires that changes in accounting for deferred tax asset valuation allowances and acquired income tax uncertainties after the measurement period be recognized as a component of provision for taxes.
We account for acquisitions of entities that include inputs and processes and have the ability to create outputs as business combinations. The tangible and identifiable intangible assets acquired and liabilities assumed in a business combination are recorded based on their estimated fair values as of the business combination date, including identifiable intangible assets which either arise from a contractual or legal right or are separable from goodwill. We base the estimated fair value of identifiable intangible assets acquired in a business combination on independent third-party valuations that use information and assumptions provided by our management, which consider our estimates of inputs and assumptions that a market participant would use. Any excess purchase price over the estimated fair value assigned to the net tangible and identifiable intangible assets acquired and liabilities assumed is recorded to goodwill. The use of alternative valuation assumptions, including estimated revenue projections, growth rates, estimated cost savings, cash flows, discount rates, estimated useful lives and probabilities surrounding the achievement of contingent milestones could result in different purchase price allocations and amortization expense in current and future periods.
In circumstances where an acquisition involves a contingent consideration arrangement that meets the definition of a liability under ASC Topic 480, Distinguishing Liabilities from Equity, we recognize a liability equal to the fair value of the contingent payments we expect to make as of the acquisition date. We remeasure this liability each reporting period and record changes in the fair value as general and administrative expense.
Transaction costs associated with acquisitions are expensed as incurred in general and administrative expenses. Results of operations and cash flows of acquired companies are included in our operating results from the date of acquisition.
Intangible assets
Amortizable intangible assets include trade names, non-compete agreements, patent licensing agreements, favorable leases, developed technology, customer relationships, and rights to develop new technology acquired as part of business combinations. Customer relationships acquired through our business combinations in 2017 are amortized on an accelerated basis, utilizing free cash flows, over periods ranging from five to 12 years. All other intangible assets subject to amortization are amortized using the straight-line method over their estimated useful lives ranging from two to 15 years. All intangible assets subject to amortization are reviewed for impairment in accordance with ASC 360, Property, Plant and Equipment.
Goodwill
In accordance with ASC 350, Intangibles-Goodwill and Other (“ASC 350”), our goodwill is not amortized but is tested for impairment on an annual basis or whenever events or changes in circumstances indicate that the carrying amount of these assets may not be recoverable. Under ASC 350, we perform annual impairment reviews of our goodwill balance during the fourth fiscal quarter or more frequently if business factors indicate. In testing for impairment, we compare the fair value of our reporting unit to its carrying value including the goodwill of that unit. If the carrying value, including goodwill, exceeds the reporting unit’s fair value, we will recognize an impairment loss for the amount by which the carrying amount exceeds the reporting unit’s fair value. The loss recognized cannot exceed the total amount of goodwill allocated to that reporting unit. We did 0t incur any goodwill impairment losses in any of the periods presented.
88
In-process research and development
Intangible assets related to in-process research and development costs (“IPR&D”) are considered to be indefinite-lived until the completion or abandonment of the associated research and development efforts. If and when development is complete, the associated assets would be deemed finite-lived and would then be amortized based on their respective estimated useful lives at that point in time. During this period, the assets will not be amortized but will be tested for impairment on an annual basis and between annual tests if we become aware of any events occurring or changes in circumstances that would indicate a reduction in the fair value of the IPR&D projects below their respective carrying amounts.
During the fourth quarter and if business factors indicate more frequently, we perform an assessment of the qualitative factors affecting the fair value of our IPR&D projects. If the fair value exceeds the carrying value, there is no impairment. Impairment losses on indefinite-lived intangible assets are recognized based solely on a comparison of the fair value of an asset to its carrying value, without consideration of any recoverability test. We have not identified any such impairment losses to date.
Leases
Under ASC 842, Leases, we determine if an arrangement is a lease at inception. Operating leases are included in operating lease assets and operating lease obligations in our consolidated balance sheets. Finance leases are included in other assets and finance lease obligations in our consolidated balance sheets.
Lease assets represent our right to use an underlying asset for the lease term and lease liabilities represent our obligation to make lease payments arising from the lease. Operating lease assets and liabilities are recognized at commencement based on the present value of lease payments over the lease term. We generally use our incremental borrowing rate based on the estimated rate of interest for collateralized borrowing over a similar term of the lease payments. The operating lease asset also includes any lease payments made and is adjusted for lease incentives. Our lease terms may include options to extend or terminate the lease which are recognized when it is reasonably certain that we will exercise that option. Leases with terms of 12 months or less are not recorded on our balance sheet. Lease expense is recognized on a straight-line basis over the lease terms, or in some cases, the useful life of the underlying asset. We account for the lease and non-lease components as a single lease component.
Property and equipment, net
Property and equipment are stated at cost less accumulated depreciation and amortization. Depreciation is computed using the straight-line method over the estimated useful lives of the assets, generally between three and seven years. Leasehold improvements are amortized using the straight-line method over the shorter of the estimated useful life of the asset or the term of the lease. Maintenance and repairs are charged to expense as incurred, and improvements and betterments are capitalized. When assets are retired or otherwise disposed of, the cost and accumulated depreciation are removed from the balance sheet and any resulting gain or loss is reflected in the consolidated statements of operations in the period realized.
The estimated useful lives of property and equipment are as follows:
| Furniture and fixtures | 7 years | ||||
| Automobiles | 7 years | ||||
| Manufacturing and Laboratory equipment | 5 years | ||||
| Computer equipment | 3 years | ||||
| Software | 3 years | ||||
| Leasehold improvements | Shorter of lease term or estimated useful life | ||||
Long-lived assets
We review long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of the assets may not be recoverable. An impairment loss is recognized when the total estimated future undiscounted cash flows expected to result from the use of the asset and its eventual disposition are less than its carrying amount. Impairment, if any, is assessed using discounted cash flows or other appropriate measures of fair value. There were 0 long-lived asset impairment losses recorded for any period presented.
89
Fair value of financial instruments
Our financial instruments consist principally of cash and cash equivalents, marketable securities, accounts payable, accrued liabilities, finance leases, and liabilities associated with business combinations. The carrying amounts of certain of these financial instruments, including cash and cash equivalents, accounts receivable, accounts payable and accrued and other current liabilities approximate their current fair value due to the relatively short-term nature of these accounts. Based on borrowing rates available to us, the carrying value of finance leases approximate their fair values. Liabilities associated with business combinations are recorded at their estimated fair value.
Revenue recognition
We recognize revenue when control of the promised goods or services is transferred to the customer in an amount that reflects the consideration it expects to be entitled to in exchange for those goods or services. All revenues are generated from contracts with customers. We utilize the following practical expedients and exemptions:
•Costs to obtain or fulfill a contract are expensed when incurred because the amortization period would have been one year or less, and
•No adjustments to promised consideration were made for financing as we expect, at contract inception, that the period between the transfer of a promised good or service and when the customer pays for that good or service will be one year or less.
Test revenue
Test revenue is comprised of testing services and sales of distributed precision oncology products.
The majority of our test revenue is generated from genetic testing, in addition to somatic testing for therapy selection and personalized cancer monitoring. These testing services provide analysis and associated interpretation of the sequencing of parts of the genome. Test orders are placed under signed requisitions or contractual agreements, and we often enter into contracts with biopharmaceutical partners, other business-to-business customers (e.g., hospitals, clinics, medical centers) and insurance companies that include pricing provisions under which such tests are billed. Billing terms are generally net 30 to 60 days.
While the transaction price of diagnostic tests is originally established either via contract or pursuant to our standard list price, we often provide concessions for tests billed to insurance carriers, and therefore the transaction price for patient insurance-billed tests is considered to be variable and revenue is recognized based on an estimate of the consideration to which we will be entitled at an amount for which it is probable that a reversal of cumulative consideration will not occur. Making these estimates requires significant judgments based upon such factors as length of payer relationship, historical payment patterns, changes in contract provisions and insurance reimbursement policies. These judgments are reviewed each reporting period and updated as necessary.
We look to transfer of control in assessing timing of recognition of revenue in connection with each performance obligation. In general, revenue in connection with the service portion of our diagnostic tests is recognized upon delivery of the underlying clinical report or when the report is made available on our web portal. Outstanding performance obligations pertaining to orders received but for which the underlying report has not been issued are generally satisfied within a 30-day period.
We also generate test revenue through the sale of our precision oncology products, which is comprised primarily of sales of our distributed research use only ("RUO ") and in vitro diagnostics ("IVD") products for therapy selection. We recognize revenue on these sales once shipment has occurred. Product sales are recorded net of discounts and other deductions. Billing terms are generally net 30 days.
Shipping and handling fees billed to customers are recorded as revenue on the consolidated statements of operations. The associated shipping and handling costs are recorded as cost of revenue.
Other revenue
Other revenue is primarily generated from pharma development services provided to biopharmaceutical companies related to companion diagnostic development as well as through collaboration agreements and genome network contracts.
90
Contracts for companion diagnostic development consist primarily of milestone-based payments along with annual fees and marked-up pass-through costs. The arrangements are treated as short-term contracts for revenue recognition purposes because they allow termination of the agreements by the customers with 30 to 120 days’ written notice without a termination penalty. Upon termination, customers are required to pay for the proportion of services provided under milestones that were in progress. We recognize revenue in an amount that reflects the consideration which we expect to receive in exchange for those goods or services. We recognize revenue over time based on the progress made toward achieving the performance obligation, utilizing both input or output methods, depending on the performance obligation, including labor hours expended, tests processed, or time elapsed, that measure our progress toward the achievement of the milestone.
We also enter into collaboration and genome network contracts. Collaboration agreements provide customers with diagnostic testing and related data aggregation reporting services that are provided over the contract term. Collaboration revenue is recognized as the data and reporting services are delivered to the customer. Genome network offerings consist of subscription services related to a proprietary software platform designed to connect patients, clinicians, advocacy organizations, researchers and therapeutic developers to accelerate the understanding, diagnosis and treatment of hereditary disease. Such services are recognized on a straight-line basis over the subscription periods. Amounts due under collaboration and genome network agreements are typically billable on net 30-day terms.
Cost of revenue
Cost of revenue reflects the aggregate costs incurred in delivering our genetic offerings and includes expenses for personnel-related costs including stock-based compensation, materials and supplies, royalties, regulatory fees, commercialization fees, equipment and infrastructure expenses associated with testing and allocated overhead including rent, equipment depreciation, information technology costs, amortization of acquired intangibles and utilities.
License Agreements
We have entered and may continue to enter into license agreements to access and utilize certain technology. We evaluate if the license agreement results in the acquisition of an asset or a business and then determine if the acquired asset has the ability to generate revenues or is subject to regulatory approval. When regulatory approval is not required, we record the license as an asset and amortize it over the estimated economic life. When regulatory approval is required, we record the amount paid as a research and development expense.
Income taxes
We use the asset and liability method of accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on the differences between the financial reporting and the tax bases of assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. A valuation allowance is provided when it is more likely than not that some portion or all of a deferred tax asset will not be realized. Significant judgment is required in determining the net valuation allowance which includes our evaluation of all available evidence including past operating results, estimates on future taxable income and acquisition-related tax assets and liabilities.
We historically established a full valuation allowance against our deferred tax assets due to the uncertainty surrounding realization of such assets. In 2020, we released approximately $112.1 million of our valuation allowance to account for acquired intangibles that support the future realization of some of our deferred tax assets. Due to the overall increase of deferred tax assets, our valuation allowance has also increased from the prior year.
Stock-based compensation
We measure stock-based payment awards made to employees and directors based on the estimated fair values of the awards and recognize the compensation expense over the requisite service period. We use the Black-Scholes option-pricing model to estimate the fair value of stock option awards and employee stock purchase plan (“ESPP”) purchases. The fair value of restricted stock unit (“RSU”) awards with time-based vesting terms is based on the grant date share price. We grant performance-based restricted stock unit (“PRSU”) awards to certain employees which vest upon the achievement of certain performance conditions, subject to the employees’ continued service relationship with us. The probability of vesting is assessed at each reporting period and compensation cost is adjusted based on this probability assessment. We recognize such compensation expense on an accelerated vesting method.
91
Stock-based compensation expense for awards without a performance condition is recognized using the straight-line method. Stock-based compensation expense is based on the value of the portion of stock-based payment awards that is ultimately expected to vest. As such, our stock-based compensation is reduced for estimated forfeitures at the date of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates.
We account for stock issued in connection with business combinations based on the fair value on the date of issuance.
Advertising
Advertising expenses are expensed as incurred. We incurred advertising expenses of $11.4 million, $9.9 million and $0.6 million during the years ended December 31, 2020, 2019 and 2018, respectively.
Comprehensive loss
Comprehensive loss is composed of two components: net loss and other comprehensive income (loss). Other comprehensive income (loss) refers to gains and losses that under U.S. GAAP are recorded as an element of stockholders’ equity, but are excluded from net loss. Our other comprehensive income (loss) consists of unrealized gains or losses on investments in available-for-sale securities.
Net loss per share
Basic net loss per share is calculated by dividing net loss by the weighted-average number of common shares outstanding during the period, without consideration of common stock equivalents. Diluted net loss per share is computed by dividing net loss by the weighted-average number of common share equivalents outstanding for the period determined using the treasury stock method. Potentially dilutive securities, consisting of preferred stock, options to purchase common stock, common stock warrants, shares of common stock pursuant to ESPP, common stock issuable in connection with our Convertible Senior Notes, RSUs and PRSUs, are considered to be common stock equivalents and were excluded from the calculation of diluted net loss per share because their effect would be antidilutive for all periods presented.
Prior period reclassifications
We have reclassified certain amounts in prior periods to conform with current presentation.
Immaterial correction of an error
We determined the historical classification of certain acquisition-related obligations as equity and the subsequent measurement of such obligations was inappropriate and instead should have been classified as liabilities and subsequently measured at fair value with changes recognized in other expense, net during the year ended December 31, 2020. We determined that the impact of the error to previously issued consolidated financial statements was not material and have corrected the immaterial error in the current period financial statements. The impact of this correction was an increase to other long-term liabilities of $10.1 million, a corresponding decrease to additional paid-in capital of $10.4 million and an increase to other income, net of $0.3 million.
Recent accounting pronouncements
We evaluate all Accounting Standards Updates (“ASUs”) issued by the Financial Accounting Standards Board ("FASB") for consideration of their applicability. ASUs not included in the disclosures in this report were assessed and determined to be either not applicable or are not expected to have a material impact on our consolidated financial statements.
Recently issued accounting pronouncements not yet adopted
In August 2020, the FASB issued ASU No. 2020-06, Accounting for Convertible Instruments and Contracts in an Entity's Own Equity, which simplifies the accounting for certain convertible instruments, amends the guidance on derivative scope exceptions for contracts in an entity's own equity, and modifies the guidance on diluted earnings per share calculations as a result of these changes. This new standard is effective for our interim and annual periods beginning January 1, 2022, and earlier adoption is permitted. We may elect to apply the amendments on a retrospective or modified retrospective basis. We are currently evaluating the impact of the adoption of this standard on our consolidated financial statements.
92
Recently adopted accounting pronouncements
In June 2016, FASB issued ASU 2016-13, Financial Instruments-Credit Losses (Topic 326), which requires measurement and recognition of expected credit losses for financial assets. This guidance became effective for us beginning in the first quarter of 2020 and was adopted using a modified retrospective approach, with certain exceptions. The adoption of Topic 326 did not have a material impact on our consolidated financial statements as credit losses are not expected to be significant.
As part of our adoption of Topic 326, we assess our accounts receivables for expected credit losses at each reporting period by disaggregating by payer type and further by portfolios of customers with similar characteristics, such as customer type and geographic location. We then review each portfolio for expected credit losses based on historical payment trends as well as forward looking data and current economic trends. If a credit loss is determined, we record a reduction to our accounts receivable balance with a corresponding general and administrative expense.
In accordance with Topic 326, we no longer evaluate whether our available-for-sale debt securities in an unrealized loss position are other than temporarily impaired. Instead, we assess whether such unrealized loss positions are credit-related. Our expected loss allowance methodology for these securities is developed by reviewing the extent of the unrealized loss, the issuers’ credit ratings and any changes in those ratings, as well as reviewing current and future economic market conditions and the issuers’ current status and financial condition. The credit-related portion of unrealized losses, and any subsequent improvements, are recorded in other income (expense), net. Unrealized gains and losses that are not credit-related are included in accumulated other comprehensive income (loss).
On January 1, 2019, we adopted the provisions of ASC Topic 842, Leases, using the modified retrospective approach in accordance with Topic 842. Adoption of Topic 842 had a material impact on our consolidated balance sheets, but did not have an impact on our consolidated statements of operations. We elected the package of practical expedients permitted under the transition guidance which, among other things, allowed us to carry forward the historical classification of leases in place as of January 1, 2019. We did not identify any material embedded leases with the adoption of Topic 842 and therefore the implementation of Topic 842 primarily focused on the treatment of our previously identified leases.
Prior period amounts were not adjusted and continue to be reported in accordance with our historic accounting under previous lease guidance, ASC 840, Leases. Under ASC 840, we rented facilities under operating lease agreements and recognized related rent expense on a straight-line basis over the term of the applicable lease agreement. Some of the lease agreements contained rent holidays, scheduled rent increases, lease incentives, and renewal options. Rent holidays and scheduled rent increases were included in the determination of rent expense recorded over the lease term. Lease incentives were recognized as a reduction of rent expense on a straight-line basis over the term of the lease. Renewals were not assumed in the determination of the lease term unless they were deemed to be reasonably assured at the inception of the lease. We recognized rent expense beginning on the date we obtained the legal right to use and control the leased space.
3. Revenue, accounts receivable and deferred revenue
Test revenue is generated from sales of diagnostic tests and precision oncology products to four groups of customers: biopharmaceutical partners; patients who pay directly; patients' insurance carriers; and other business-to-business customers (e.g., hospitals, clinics, medical centers). Test revenue is generated in two ways: through a centralized lab and decentralized through the shipment of reactions to biopharma partners and other business-to-business customers. We refer to the set of reagents needed to perform a next-generation sequencing test as a "reaction." Amounts billed and collected, and the timing of collections, vary based on the type of payer. Other revenue consists principally of revenue recognized under contracts for pharma development services and other collaboration and genome network agreements and is accounted for under the provisions provided in ASC 606, Revenue from Contracts with Customers.
93
Our revenue as disaggregated by payer category and revenue subtype is as follows (in thousands):
| Patient | Biopharma partner | Other business-to-business | Year Ended December 31, 2020 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Insurance | Direct | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Test revenue: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Centralized | $ | 181,026 | $ | 23,972 | $ | 26,228 | $ | 32,736 | $ | 263,962 | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Decentralized | 0 | 0 | 837 | 7,511 | 8,348 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Total test revenue | 181,026 | 23,972 | 27,065 | 40,247 | 272,310 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Other revenue | 0 | 0 | 4,488 | 2,800 | 7,288 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Total revenue | $ | 181,026 | $ | 23,972 | $ | 31,553 | $ | 43,047 | $ | 279,598 | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Patient | Biopharma partner | Other business-to-business | Year Ended December 31, 2019 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Insurance | Direct | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Test revenue: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Centralized | $ | 153,827 | $ | 17,597 | $ | 10,876 | $ | 30,173 | $ | 212,473 | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Total test revenue | 153,827 | 17,597 | 10,876 | 30,173 | 212,473 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Other revenue | 0 | 0 | 2,077 | 2,274 | 4,351 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Total revenue | $ | 153,827 | $ | 17,597 | $ | 12,953 | $ | 32,447 | $ | 216,824 | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Patient | Biopharma partner | Other business-to-business | Year Ended December 31, 2018 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Insurance | Direct | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Test revenue: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Centralized | $ | 96,352 | $ | 13,589 | $ | 6,231 | $ | 28,388 | $ | 144,560 | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Total test revenue | 96,352 | 13,589 | 6,231 | 28,388 | 144,560 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Other revenue | 0 | 0 | 1,565 | 1,574 | 3,139 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Total revenue | $ | 96,352 | $ | 13,589 | $ | 7,796 | $ | 29,962 | $ | 147,699 | ||||||||||||||||||||||||||||||||||||||||||||||||||||
We recognize revenue related to billings based on estimates of the amount that will ultimately be realized. Cash collections for certain tests delivered may differ from rates originally estimated. As a result of new information, we update our estimate quarterly of the amounts to be recognized for previously delivered tests which resulted in the following increases to revenue and decreases to our loss from operations and basic and diluted net loss per share (in millions, except per share amounts):
| Year Ended December 31, | |||||||||||||||||
| 2020 | 2019 | 2018 | |||||||||||||||
| Revenue | $ | 4.4 | $ | 4.1 | $ | 4.5 | |||||||||||
| Loss from operations | $ | (4.4) | $ | (4.1) | $ | (4.5) | |||||||||||
| Net loss per share, basic and diluted | $ | (0.03) | $ | (0.05) | $ | (0.07) | |||||||||||
Impact of COVID-19
Our billable volumes decreased significantly in the second half of March 2020 as compared to the first few months of 2020 as a result of COVID-19 and related limitations and priorities across the healthcare system. Our daily test volumes have consistently increased from the low in March 2020, although we are currently still experiencing changes in product mix due to the impact of COVID-19. COVID-19 could have a material impact on our financial results for the foreseeable future, particularly on product mix and as a result, the revenue we recognize. We have reviewed and adjusted for the impact of COVID-19 on our estimates related to revenue recognition and expected credit losses.
Approximately 8% of our workforce as of March 31, 2020 was impacted by a reduction in force in April 2020 in an initiative to manage costs and cash burn that resulted in one-time costs in the second quarter of 2020 of $3.8 million. In addition, effective May 2020, we have reduced the salaries of our named executive officers by approximately 20%, which reductions ceased as of January 2021.
94
On March 27, 2020, the Coronavirus Aid, Relief, and Economic Security Act ("CARES Act") was signed into law which was a stimulus bill intended to bolster the economy, among other things, and provide assistance to qualifying businesses and individuals. The CARES Act included an infusion of funds into the healthcare system, and in April 2020, we received $3.8 million as a part of this initiative. This payment was recognized as other income (expense), net in our consolidated statement of operations during the year ended December 31, 2020. We also received $2.3 million in January 2021 which we recognized as other income (expense), net during the three months ended March 31, 2021. At this time, we are not certain of the availability, extent or impact of any future relief provided under the CARES Act.
Accounts receivable
The majority of our accounts receivable represents amounts billed to pharmaceutical partners and other business-to-business customers for test and other revenue recognized, and estimated amounts to be collected from third-party insurance payers for genetic testing revenue recognized. Also included are amounts due under the terms of collaboration and genome network agreements for diagnostic testing and data aggregation reporting services provided and proprietary platform access rights transferred.
We also record unbilled revenue for revenue recognized but yet to be billed for services provided to biopharmaceutical companies related to companion diagnostic development. The amount is a contract receivable and is included in accounts receivable on the consolidated balance sheets; unbilled revenue was $4.3 million and NaN as of December 31, 2020 and 2019, respectively.
Deferred revenue
We record a contract liability when cash payments are received or due in advance of our performance related to one or more performance obligations. The deferred revenue balance primarily consists of advanced billings for pharma development services, including billings at the initiation of a performance-based milestones, and recognized as revenue in the applicable future period when the revenue is earned. Also included are prepayments related to our consumer direct channel. We recognized revenue of $1.4 million from deferred revenue for the year ended as of December 31, 2020. In addition, we recognized deferred revenue of $4.8 million upon the acquisition of ArcherDX in October 2020, $2.0 million of which was recognized as revenue during the year ended December 31, 2020.
4. Business combinations
Singular Bio
In June 2019, we acquired 100% of the fully diluted equity of Singular Bio, a privately held company developing single molecule detection technology, for approximately $57.3 million, comprised of $53.9 million in the form of 2.5 million shares of our common stock and the remainder in cash.
We granted approximately $90.0 million of restricted stock units ("RSU") under our 2015 Stock Incentive Plan as inducement awards to new employees who joined Invitae in connection with our acquisition of Singular Bio. $45.0 million of the RSUs are time-based and vested in three equal installments in December 2019, June 2020, and December 2020, subject to the employee's continued service with us ("Time-based RSUs") and $45.0 million of the RSUs are performance-based RSUs ("PRSU") that vest upon the achievement of certain performance conditions. Since the number of awards granted is based on a 30-day volume weighted-average share price with a fixed dollar value, these Time-based RSUs and PRSUs are liability-classified and the fair value is estimated at each reporting period based on the number of shares that are expected to be issued at each reporting date and our closing stock price, which combined are categorized as Level 3 inputs. Therefore, fair value of the RSUs and PRSUs and the number of shares to be issued will not be fixed until the awards vest.
During the years ended December 31, 2020 and 2019, we recorded research and development stock-based compensation expense of $29.1 million and $14.7 million, respectively, related to the Time-based RSUs, and $19.4 million and $24.4 million, respectively, related to the PRSUs based on our evaluations of the probability of achieving performance conditions. As of December 31, 2020, the Time-based RSUs and PRSUs had a total fair value of $43.9 million and $43.8 million, respectively, based on a total estimated issuance of 3.5 million shares and expectation of the achievement of the performance conditions. As of December 31, 2020, 2.0 million of the Time-based RSUs and 1.2 million of the PRSUs had vested with a total fair value of $75.0 million which was recorded in common stock issued or issuable pursuant to acquisitions in the consolidated statements of stockholders' equity.
95
Jungla
In July 2019, we acquired 100% of the equity interest of Jungla, a privately held company developing a platform for molecular evidence testing in genes, for approximately $59.0 million, comprised of $44.9 million in the form of shares of our common stock and the remainder in cash.
We may be required to pay contingent consideration based on achievement of post-closing development milestones. As of the acquisition date, the fair value of this contingent consideration was $10.7 million including cash and common stock. These milestones are expected to be completed within two years of the date of acquisition, two of which were completed during the year ended December 31, 2020. The material factors that may impact the fair value of the contingent consideration, and therefore, this liability, are the probabilities and timing of achieving the related milestones and the discount rate we used to estimate the fair value, which are Level 3 inputs not supported by market activity. Significant changes in any of the probabilities of success would result in a significant change in the fair value, which will be estimated at each reporting date with changes reflected as a general and administrative expense. As of December 31, 2020, the fair value of this contingent consideration was $7.1 million.
Upon acquisition, we had a stock payable liability related to our acquisition of Jungla which represents the hold-back obligation to issue 0.2 million shares subject to indemnification claims that may arise. This liability was adjusted at each reporting period based on the fair value of our common stock, which is a Level 3 input, with the change recorded in other income (expense), net. During July 2020, the hold-back shares were remitted in full to the former owners of Jungla.
Clear Genetics
In November 2019, we acquired 100% of the equity interest of Clear Genetics, a developer of software for providing genetic services at scale, for approximately $50.1 million. Of the cash and stock purchase price consideration issued, $0.2 million of cash and approximately 0.4 million shares of our common stock were subject to a 12-month hold back to satisfy indemnification obligations that were released during the year ended December 31, 2020.
Diploid
In March 2020, we acquired 100% of the equity interest of Diploid, a developer of artificial intelligence software capable of diagnosing genetic disorders using sequencing data and patient information, for approximately $82.3 million in cash and shares of our common stock. Of the stock purchase price consideration issued, approximately 0.4 million shares are subject to a hold-back to satisfy indemnification obligations that may arise. We included the financial results of Diploid in our consolidated financial statements from the acquisition date, which were not material for the year ended December 31, 2020.
96
The following table summarizes the purchase price recorded as a part of the acquisition of Diploid (in thousands):
| Purchase Price | |||||
| Cash transferred | $ | 32,323 | |||
| Hold-back consideration - common stock | 7,538 | ||||
| Common stock transferred | 42,453 | ||||
| Total | $ | 82,314 | |||
Assets acquired and liabilities assumed are recorded based on valuations derived from estimated fair value assessments and assumptions used by us. While we believe that our estimates and assumptions underlying the valuations are reasonable, different estimates and assumptions could result in different valuations assigned to the individual assets acquired and liabilities assumed, and the resulting amount of goodwill. The following table summarizes the fair values of assets acquired and liabilities assumed through our acquisition of Diploid at the date of acquisition (in thousands):
| Cash | $ | 124 | |||
| Accounts receivable | 26 | ||||
| Developed technology | 41,789 | ||||
| Total identifiable assets acquired | 41,939 | ||||
| Accounts payable | (30) | ||||
| Deferred tax liability | (10,250) | ||||
| Net identifiable assets acquired | 31,659 | ||||
| Goodwill | 50,655 | ||||
| Total purchase price | $ | 82,314 | |||
Based on the guidance provided in ASC 805, Business Combinations, we accounted for the acquisition of Diploid as a business combination and determined that 1) Diploid was a business which combines inputs and processes to create outputs, and 2) substantially all of the fair value of gross assets acquired was not concentrated in a single identifiable asset or group of similar identifiable assets.
Our purchase price allocation for the acquisition is preliminary and subject to revision as additional information about fair value of assets and liabilities becomes available, primarily related to our deferred tax liability assumed in connection with the acquisition. Additional information that existed as of the acquisition date but at the time was unknown to us may become known to us during the remainder of the measurement period, a period not to exceed 12 months from the acquisition date.
We measured the identifiable assets and liabilities assumed at their acquisition date fair values separately from goodwill. The intangible asset acquired is developed technology related to Diploid's artificial intelligence technology platform. The fair value of the developed technology was estimated using an income approach with an estimated useful life of nine years. As of the acquisition date, we recorded a stock payable liability of $7.5 million to represent the hold-back obligation to issue 0.4 million shares subject to indemnification claims that may arise. This liability is adjusted at each reporting period based on the fair value of our common stock, which is a Level 3 input. As of December 31, 2020, the value of this liability was $17.7 million with the change recorded in other income (expense), net.
Goodwill represents the excess of the purchase price over the fair value of the net tangible and intangible assets acquired. The acquisition of Diploid resulted in the recognition of $50.7 million of goodwill which we believe relates primarily to expansion of the acquired technology to apply to new areas of genetic testing. The goodwill created as a result of the acquisition of Diploid is not deductible for the foreign local tax purposes.
In June 2020, we granted 0.2 million RSUs with a fair value of $3.6 million under our 2015 Stock Incentive Plan as inducement awards in connection with our acquisition of Diploid. These RSUs vest in two equal installments, in April 2021 and April 2022. The value of the awards was recognized as research and development stock-based compensation upon grant in June 2020 as there were no ongoing obligations required by the award recipients.
Genelex and YouScript
In April 2020, we acquired 100% of the equity interest of Genelex and YouScript to bring pharmacogenetic testing and integrated clinical decision support to Invitae. We acquired Genelex for approximately $13.2 million,
97
primarily in shares of our common stock. Of the stock purchase price consideration issued, approximately 0.1 million shares are subject to a hold-back to satisfy indemnification obligations that may arise. We acquired YouScript for approximately $52.7 million, including cash consideration of $24.5 million and the remaining in shares of our common stock. Of the purchase price consideration for YouScript, approximately $1.4 million and 0.5 million shares of our common stock are subject to a hold-back to satisfy indemnification obligations that may arise. We included the financial results of Genelex and YouScript in our consolidated financial statements from the acquisition date, which were not material for the year ended December 31, 2020. We recorded $1.7 million of transaction costs related to the acquisition of Genelex and YouScript as general and administrative expense during the year ended December 31, 2020.
We may be required to pay contingent consideration in the form of additional shares of our common stock in connection with the acquisition of Genelex if, within a specified period following the closing, we achieve a certain product milestone, in which case we would issue shares of our common stock with a value equal to a portion of the gross revenues actually received by us for a pharmacogenetic product reimbursed through certain payers during an earn-out period of up to four years. As of the acquisition date, the fair value of this contingent consideration was $2.0 million in the form of shares of our common stock. The material factors that may impact the fair value of the contingent consideration, and therefore, this liability, are the probabilities and timing of achieving the related milestone, the estimated revenues achieved for a pharmacogenetic product and the discount rate we used to estimate the fair value, which are Level 3 inputs not supported by market activity. Significant changes in any of the probabilities of success would result in a significant change in the fair value, which is estimated at each reporting date with changes reflected as general and administrative expense. As of December 31, 2020, the fair value of this contingent consideration was $1.2 million.
The following table summarizes the purchase prices recorded as a part of the acquisition of Genelex and YouScript (in thousands):
| Genelex | YouScript | Total | |||||||||||||||
| Cash transferred | $ | 972 | $ | 24,462 | $ | 25,434 | |||||||||||
| Hold-back consideration - cash | 0 | 1,385 | 1,385 | ||||||||||||||
| Hold-back consideration - common stock | 781 | 5,392 | 6,173 | ||||||||||||||
| Contingent consideration | 1,994 | 0 | 1,994 | ||||||||||||||
| Common stock transferred | 9,463 | 21,464 | 30,927 | ||||||||||||||
| Total | $ | 13,210 | $ | 52,703 | $ | 65,913 | |||||||||||
98
Assets acquired and liabilities assumed are recorded based on valuations derived from estimated fair value assessments and assumptions used by us. While we believe that our estimates and assumptions underlying the valuations are reasonable, different estimates and assumptions could result in different valuations assigned to the individual assets acquired and liabilities assumed, and the resulting amount of goodwill. The following table summarizes the fair values of assets acquired and liabilities assumed through our acquisitions of Genelex and YouScript at the date of acquisition (in thousands):
| Genelex | YouScript | Total | |||||||||||||||
| Cash | $ | 33 | $ | 24 | $ | 57 | |||||||||||
| Accounts receivable | 221 | 56 | 277 | ||||||||||||||
| Prepaid expenses and other current assets | 0 | 70 | 70 | ||||||||||||||
| Operating lease assets | 0 | 355 | 355 | ||||||||||||||
| Developed technology | 9,209 | 25,716 | 34,925 | ||||||||||||||
| Total identifiable assets acquired | 9,463 | 26,221 | 35,684 | ||||||||||||||
| Current liabilities | (320) | (481) | (801) | ||||||||||||||
| Deferred tax liability | 0 | (2,600) | (2,600) | ||||||||||||||
| Other long-term liabilities | 0 | (163) | (163) | ||||||||||||||
| Net identifiable assets acquired | 9,143 | 22,977 | 32,120 | ||||||||||||||
| Goodwill | 4,067 | 29,726 | 33,793 | ||||||||||||||
| Total purchase price | $ | 13,210 | $ | 52,703 | $ | 65,913 | |||||||||||
Based on the guidance provided in ASC 805, Business Combinations, we accounted for the acquisitions of Genelex and YouScript as business combinations and determined that 1) Genelex and YouScript were businesses which combine inputs and processes to create outputs, and 2) substantially all of the fair value of gross assets acquired were not concentrated in a single identifiable asset or group of similar identifiable assets.
Our purchase price allocation for the acquisitions is preliminary and subject to revision as additional information about fair value of assets and liabilities becomes available, primarily related to our deferred tax liability assumed. Additional information that existed as of the acquisition date but at the time was unknown to us may become known to us during the remainder of the measurement period, a period not to exceed 12 months from the acquisition date.
We measured the identifiable assets and liabilities assumed at their acquisition date fair values separately from goodwill. The intangible assets acquired are the developed technologies related to Genelex's and YouScript's technology platforms. The fair value of the developed technologies were estimated using an income approach with an estimated useful life of eight years. As of the acquisition date, we recorded stock payable liabilities of $6.2 million to represent the hold-back obligation to issue shares subject to indemnification claims that may arise. These liabilities are adjusted at each reporting period based on the fair value of our common stock, which is a Level 3 input. As of December 31, 2020, the value of this liability was $21.6 million with the change recorded in other income (expense), net.
Goodwill represents the excess of the purchase price over the fair value of the net tangible and intangible assets acquired. The acquisitions of Genelex and YouScript resulted in the recognition of $33.8 million of goodwill which we believe relates primarily to future functionality and expansion of the acquired technologies. Of the goodwill recognized, $29.7 million related to the YouScript acquisition is not deductible for tax purposes.
ArcherDX
In June 2020, we entered into a definitive agreement with ArcherDX, a genomics analysis company democratizing precision oncology, and in October 2020, the closing conditions were met and the transaction was consummated. Under the terms of the agreement, we acquired 100% of the equity interest of ArcherDX for $2.3 billion, comprised of $2.0 billion in the form of our common stock, $2.0 million in liabilities, and the remainder in cash. We incurred $20.9 million of transaction costs related to the acquisition of ArcherDX which we recorded as general and administrative expense during the year ended December 31, 2020.
99
We may be required to pay contingent consideration based on achievement of post-closing development and revenue milestones. As of the acquisition date, the total fair value of the contingent consideration was $945.2 million, $933.6 million of which was included in the purchase price and $11.6 million recognized as non-recurring post-combination compensation expense. Of this $933.6 million, $925.1 million would be in the form of shares of our common stock which will be priced at the time of the milestone achievement, and the remainder in cash. The milestones are expected to be completed within approximately two years from the date of the acquisition, with one of them being achieved during November 2020 which resulted in the issuance of 5.0 million shares of our common stock and a cash payment of $1.9 million. This milestone achievement subsequent to the acquisition date resulted in the recognition of $40.6 million general and administrative expense. The material factors that may impact the fair value of the contingent consideration, and therefore the liability, are (i) the estimated number of shares issued, (ii) the volatility assumptions of our common stock used in the Monte Carlo simulation, (iii) the probabilities and timing of achievement of milestones and (iv) discount rates, all of which are Level 3 inputs not supported by market activity. Significant changes in any of these inputs may result in a significant change in fair value, which is estimated at each reporting date with changes reflected as general and administrative expense. As of December 31, 2020, the fair value of the contingent consideration representing the remaining milestones was $788.3 million.
In connection with the acquisition, all of ArcherDX's equity awards outstanding and unvested prior to the acquisition became fully vested per the terms of the agreement. The acceleration of vesting required us to allocate the fair value of the equity attributable to the pre-combination service to the purchase price and the remaining amount of $125.8 million, inclusive of $11.6 million in contingent consideration, to non-recurring post-combination expense which we recognized as general and administrative expense during the year ended December 31, 2020.
We included the financial results of ArcherDX in our consolidated financial statements from the acquisition date, which contributed $16.2 million and $24.8 million of revenue and net loss, respectively, during the year ended December 31, 2020.
The following table summarizes the purchase price and post-combination expense recorded as a part of the acquisition of ArcherDX (in millions):
| Purchase Price | Post-combination Expense | ||||||||||
| Cash transferred | $ | 335.3 | $ | 22.5 | |||||||
| Contingent consideration and liabilities incurred | 935.6 | 12.3 | |||||||||
| Common stock transferred | 1,060.6 | 91.0 | |||||||||
| Total | $ | 2,331.5 | $ | 125.8 | |||||||
100
Assets acquired and liabilities assumed are recorded based on valuations derived from estimated fair value assessments and assumptions used by us. While we believe that our estimates and assumptions underlying the valuations are reasonable, different estimates and assumptions could result in different valuations assigned to the individual assets acquired and liabilities assumed, and the resulting amount of goodwill. The following table summarizes the fair values of assets acquired and liabilities assumed through our acquisition of ArcherDX at the date of acquisition (in millions):
| Cash | $ | 9.1 | |||
| Accounts receivable | 12.1 | ||||
| Inventory | 17.6 | ||||
| Prepaid expenses and other current assets | 6.8 | ||||
| Property and equipment, net | 17.1 | ||||
| Operating lease assets | 7.9 | ||||
| Intangible assets | 803.8 | ||||
| Other assets | 0.7 | ||||
| Total identifiable assets acquired | 875.1 | ||||
| Accounts payable | (4.6) | ||||
| Accrued liabilities | (18.0) | ||||
| Operating lease obligations | (1.3) | ||||
| Operating lease obligations, net of current portion | (7.4) | ||||
| Deferred tax liability | (151.1) | ||||
| Other liabilities | (13.6) | ||||
| Net identifiable assets acquired | 679.1 | ||||
| Goodwill | 1,652.4 | ||||
| Total purchase price | $ | 2,331.5 | |||
Based on the guidance provided in ASC 805, Business Combinations, we accounted for the acquisition of ArcherDX as a business combination and determined that 1) ArcherDX was a business which combines inputs and processes to create outputs, and 2) substantially all of the fair value of gross assets acquired was not concentrated in a single identifiable asset or group of similar identifiable assets.
Our purchase price allocation for the acquisition is preliminary and subject to revision as additional information about fair value of assets and liabilities becomes available, primarily related to our deferred tax liability assumed in connection with the acquisition. Additional information that existed as of the acquisition date but at the time was unknown to us may become known to us during the remainder of the measurement period, a period not to exceed 12 months from the acquisition date.
101
We measured the identifiable assets and liabilities assumed at their acquisition date fair values separately from goodwill. The intangible assets acquired are the developed technology related to ArcherDX's artificial intelligence technology platform, IPR&D for its STRATAFIDE and PCM products, ArcherDX's customer relationships in place at the time of acquisition, and the ArcherDX tradename. We also acquired the right to develop new technology through an existing agreement for the development and commercialization of sequencing-based companion diagnostics between ArcherDX and a vendor. The fair value of our intangible assets acquired as of the acquisition date and the method used to value these assets as well as the estimated economic lives for amortizable intangible assets were as follows (in millions, except estimated useful life which is in years):
| Fair value | Estimated useful life | Valuation method | Amortization expense | ||||||||||||||||||||
| Customer relationships | $ | 17.3 | 12.0 | With-and-without | Selling and marketing | ||||||||||||||||||
| Tradename | 21.1 | 12.0 | Relief from royalty | Selling and marketing | |||||||||||||||||||
| Developed technology | 233.6 | 12.0 | Multi-period excess earnings | Cost of revenue | |||||||||||||||||||
| Right to develop new technology | 19.4 | 15.0 | Cost approach | Research and development | |||||||||||||||||||
| In-process research and development | 512.4 | n/a | Multi-period excess earnings | Not applicable | |||||||||||||||||||
| Total | $ | 803.8 | |||||||||||||||||||||
Goodwill represents the excess of the purchase price over the fair value of the net tangible and intangible assets acquired. The acquisition of ArcherDX resulted in the recognition of $1.7 billion of goodwill which we believe relates primarily to the anticipated benefits of synergies created through the acquisition and assembled workforce. The acquisition of ArcherDX advances our objectives to create a comprehensive offering that provides testing services for disease risk, therapy selection and personalized cancer monitoring to enable precision approaches to cancer treatment. Goodwill created as a result of the acquisition of ArcherDX is not deductible for tax purposes.
We recorded an income tax benefit of $109.5 million in the three months ended December 31, 2020 due to net deferred tax liabilities assumed in connection with our acquisition of ArcherDX which provided a future source of income to support the realization of our deferred tax assets and resulted in a partial release of our valuation allowance.
In connection with the acquisition, we granted inducement awards of Invitae common stock to new employees who joined Invitae in connection with our acquisition of ArcherDX with an estimated fair value of $112.2 million, net of estimated forfeitures. These awards vest upon the achievement of the contingent consideration milestones discussed above and are subject to the employee’s continued service with us, unless terminated without cause in which case vesting is only dependent on milestone achievement. As the number of shares that are expected to be issued are fixed, the awards are equity-classified. During the year ended December 31, 2020, we recorded $41.2 million in stock-based compensation expense based on our probability of milestone achievement. Included in the stock-based compensation expense is $2.1 million related to the acceleration of stock-based compensation expense due to the termination of an employee without cause whereby the employee's continued service is not required.
Pro forma financial information (unaudited)
The audited pro forma financial information in the table below summarizes the combined results of operations for Invitae and ArcherDX as though the companies had been combined as of January 1, 2019. The pro forma amounts have been adjusted for:
•transaction expenses incurred by ArcherDX and us,
•depreciation expense resulting from the fair value of the acquired property and equipment,
•amortization expense resulting from the acquired intangible assets,
•the elimination of historical interest expense incurred by ArcherDX on its debt and debt-like items and the incurrence of interest expense related to the issuance of debt in connection with the acquisition,
•lease expense resulting from the step-up in the operating lease obligation and operating lease asset,
•nonrecurring post-combination expense,
•income tax benefits resulting from the deferred tax liabilities acquired, and
•the 26.3 million shares of our common stock issued upon the closing of the ArcherDX transaction.
102
The following unaudited pro forma financial information is for informational purposes only and is not necessarily indicative of the results of operations that would have been achieved as if the acquisition had taken place as of January 1, 2019 (in thousands):
| Year Ended December 31, | ||||||||||||||||||||||||||||||||||||||
| 2020 | 2019 | |||||||||||||||||||||||||||||||||||||
| Revenue | $ | 327,233 | $ | 267,389 | ||||||||||||||||||||||||||||||||||
| Net loss | (685,589) | (355,818) | ||||||||||||||||||||||||||||||||||||
5. Goodwill and intangible assets
Goodwill
The changes in the carrying amounts of goodwill were as follows (in thousands):
| Balance as of December 31, 2019 | $ | 126,777 | ||||||
| Goodwill acquired - Diploid | 50,655 | |||||||
| Goodwill acquired - Genelex | 4,067 | |||||||
| Goodwill acquired - YouScript | 29,726 | |||||||
| Goodwill acquired - ArcherDX | 1,652,398 | |||||||
| Balance as of December 31, 2020 | $ | 1,863,623 | ||||||
Intangible assets
The following table presents details of our acquired intangible assets as of December 31, 2020 (in thousands):
| Cost | Accumulated Amortization | Net | Weighted-Average Useful Life (in Years) | Weighted-Average Estimated Remaining Useful Life (in Years) | |||||||||||||||||||||||||
| Customer relationships | $ | 41,075 | $ | (8,292) | $ | 32,783 | 10.8 | 8.8 | |||||||||||||||||||||
| Developed technology | 397,563 | (31,013) | 366,550 | 10.6 | 10.0 | ||||||||||||||||||||||||
| Non-compete agreement | 286 | (229) | 57 | 5.0 | 1.0 | ||||||||||||||||||||||||
| Tradename | 21,085 | (447) | 20,638 | 12.0 | 11.8 | ||||||||||||||||||||||||
| Patent licensing agreement | 496 | (103) | 393 | 15.0 | 11.9 | ||||||||||||||||||||||||
| Right to develop new technology | 19,359 | (323) | 19,036 | 15.0 | 14.8 | ||||||||||||||||||||||||
| In-process research and development | 542,388 | — | 542,388 | n/a | n/a | ||||||||||||||||||||||||
| $ | 1,022,252 | $ | (40,407) | $ | 981,845 | 10.9 | 10.2 | ||||||||||||||||||||||
Acquisition-related intangibles included in the above table are finite-lived, other than in-process research and development which has an indefinite life, and are carried at cost less accumulated amortization. Customer relationships related to our 2017 business combinations are being amortized on an accelerated basis, in proportion to estimated cash flows. All other finite-lived acquisition-related intangibles are being amortized on a straight-line basis over their estimated lives, which approximates the pattern in which the economic benefits of the intangible assets are expected to be realized. Amortization expense was $26.6 million, $7.7 million, and $5.0 million for the years ended December 31, 2020, 2019 and 2018, respectively. Intangible assets are carried at cost less accumulated amortization. Amortization expense is recorded to cost of revenue, research and development, sales and marketing and general and administrative expense.
The following table summarizes our estimated future amortization expense of intangible assets with finite lives as of December 31, 2020 (in thousands):
| 2021 | $ | 46,910 | |||
| 2022 | 45,401 | ||||
| 2023 | 44,388 | ||||
| 2024 | 44,110 | ||||
| 2025 | 42,356 | ||||
| Thereafter | 216,292 | ||||
| Total estimated future amortization expense | $ | 439,457 | |||
103
In December 2020, we entered into an agreement to acquire technology focused on informing clinical decisions for $2.9 million. We accounted for this transaction as an asset acquisition of developed technology which will be amortized over eight years, initially to research and development expense. In connection with this transaction, we granted approximately $6.2 million of RSUs under our 2015 Stock Incentive Plan as inducement awards to new employees who joined Invitae. These RSUs are time-based and vest in two equal installments in December 2021 and December 2022, subject to the employee's continued service with us. For $5.4 million of these awards, the number of awards granted are based on the lower of the 20-day volume weighted-average share price prior to the vesting date and the date of close, both with a fixed dollar value, and therefore, these RSUs are liability-classified and the fair value is estimated at each reporting period based on the number of shares that are expected to be issued at each reporting date and our closing stock price, which combined are categorized as Level 3 inputs. Therefore, fair value of these RSUs and the number of shares to be issued will not be fixed until the awards vest. During the year ended December 31, 2020, we recorded research and development stock-based compensation expense of $0.2 million related to the RSUs based on an estimated issuance of 0.1 million shares.
6. Balance sheet components
Inventory
Inventory consisted of the following (in thousands):
| December 31, | |||||||||||
| 2020 | 2019 | ||||||||||
| Raw materials | $ | 21,324 | $ | 6,569 | |||||||
| Work in progress | 8,847 | 79 | |||||||||
| Finished goods | 1,859 | 0 | |||||||||
| Total inventory | $ | 32,030 | $ | 6,648 | |||||||
Property and equipment, net
Property and equipment consisted of the following (in thousands):
| December 31, | |||||||||||
| 2020 | 2019 | ||||||||||
| Leasehold improvements | $ | 26,516 | $ | 18,352 | |||||||
| Laboratory equipment | 45,342 | 24,873 | |||||||||
| Computer equipment | 10,939 | 5,995 | |||||||||
| Software | 566 | 2,611 | |||||||||
| Furniture and fixtures | 1,967 | 1,198 | |||||||||
| Automobiles | 58 | 58 | |||||||||
| Construction-in-progress | 12,061 | 10,795 | |||||||||
| Total property and equipment, gross | 97,449 | 63,882 | |||||||||
| Accumulated depreciation and amortization | (31,347) | (26,135) | |||||||||
| Total property and equipment, net | $ | 66,102 | $ | 37,747 | |||||||
Depreciation expense was $10.5 million, $7.1 million and $8.5 million for the years ended December 31, 2020, 2019 and 2018, respectively.
104
Accrued liabilities
Accrued liabilities consisted of the following (in thousands):
| December 31, | |||||||||||
| 2020 | 2019 | ||||||||||
| Accrued compensation and related expenses | $ | 25,221 | $ | 16,440 | |||||||
| Deferred revenue | 6,378 | 1,429 | |||||||||
| Compensation and other liabilities associated with business combinations | 25,600 | 30,560 | |||||||||
| Other | 28,859 | 16,385 | |||||||||
| Total accrued liabilities | $ | 86,058 | $ | 64,814 | |||||||
Other long-term liabilities
Other long-term liabilities consisted of the following (in thousands):
| December 31, | |||||||||||
| 2020 | 2019 | ||||||||||
| Deferred revenue, non-current | 1,380 | 0 | |||||||||
| Compensation and other liabilities associated with business combinations, non-current | 825,976 | 8,000 | |||||||||
| Other | 13,900 | 0 | |||||||||
| Total other long-term liabilities | $ | 841,256 | $ | 8,000 | |||||||
7. Fair value measurements
Financial assets and liabilities are recorded at fair value. Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability (an exit price) in an orderly transaction between market participants at the reporting date. The authoritative guidance establishes a three-level valuation hierarchy that prioritizes the inputs to valuation techniques used to measure fair value based upon whether such inputs are observable or unobservable. Observable inputs reflect market data obtained from independent sources, while unobservable inputs reflect market assumptions made by the reporting entity.
The three-level hierarchy for the inputs to valuation techniques is summarized as follows:
Level 1—Observable inputs such as quoted prices (unadjusted) for identical instruments in active markets.
Level 2—Observable inputs such as quoted prices for similar instruments in active markets, quoted prices for identical or similar instruments in markets that are not active, or model-derived valuations whose significant inputs are observable.
Level 3—Unobservable inputs that reflect the reporting entity’s own assumptions.
105
The following tables set forth the fair value of our consolidated financial instruments that were measured at fair value on a recurring basis (in thousands):
| December 31, 2020 | ||||||||||||||||||||||||||||||||||||||||||||
| Amortized Cost | Gross Unrealized Gains | Gross Unrealized Losses | Estimated Fair Value | Level 1 | Level 2 | Level 3 | ||||||||||||||||||||||||||||||||||||||
| Financial assets: | ||||||||||||||||||||||||||||||||||||||||||||
| Money market funds | $ | 83,109 | $ | 0 | $ | 0 | $ | 83,109 | $ | 83,109 | $ | 0 | $ | 0 | ||||||||||||||||||||||||||||||
| U.S. Treasury notes | 164,894 | 7 | (15) | 164,886 | 164,886 | 0 | 0 | |||||||||||||||||||||||||||||||||||||
| U.S. government agency securities | 64,291 | 9 | 0 | 64,300 | 0 | 64,300 | 0 | |||||||||||||||||||||||||||||||||||||
| Total financial assets | $ | 312,294 | $ | 16 | $ | (15) | $ | 312,295 | $ | 247,995 | $ | 64,300 | $ | 0 | ||||||||||||||||||||||||||||||
| Financial liabilities: | ||||||||||||||||||||||||||||||||||||||||||||
| Stock payable liability | $ | 39,237 | $ | 0 | $ | 0 | $ | 39,237 | ||||||||||||||||||||||||||||||||||||
| Contingent consideration | 796,639 | 0 | 0 | 796,639 | ||||||||||||||||||||||||||||||||||||||||
| Total financial liabilities | $ | 835,876 | $ | — | $ | 0 | $ | 835,876 | ||||||||||||||||||||||||||||||||||||
| December 31, 2020 | ||||||||||||||||||||||||||||||||||||||||||||
| Reported as: | ||||||||||||||||||||||||||||||||||||||||||||
| Cash equivalents | $ | 76,423 | ||||||||||||||||||||||||||||||||||||||||||
| Restricted cash | 6,686 | |||||||||||||||||||||||||||||||||||||||||||
| Marketable securities | 229,186 | |||||||||||||||||||||||||||||||||||||||||||
| Total cash equivalents, restricted cash, and marketable securities | $ | 312,295 | ||||||||||||||||||||||||||||||||||||||||||
| Accrued liabilities | $ | 10,592 | ||||||||||||||||||||||||||||||||||||||||||
| Other long-term liabilities | $ | 825,284 | ||||||||||||||||||||||||||||||||||||||||||
106
| December 31, 2019 | ||||||||||||||||||||||||||||||||||||||||||||
| Amortized Cost | Gross Unrealized Gains | Gross Unrealized Losses | Estimated Fair Value | Level 1 | Level 2 | Level 3 | ||||||||||||||||||||||||||||||||||||||
| Financial assets: | ||||||||||||||||||||||||||||||||||||||||||||
| Money market funds | $ | 39,396 | $ | 0 | $ | 0 | $ | 39,396 | $ | 39,396 | $ | 0 | $ | 0 | ||||||||||||||||||||||||||||||
| Certificates of deposit | 300 | 0 | 0 | 300 | 0 | 300 | 0 | |||||||||||||||||||||||||||||||||||||
| U.S. Treasury notes | 150,627 | 0 | (15) | 150,612 | 150,612 | 0 | 0 | |||||||||||||||||||||||||||||||||||||
| U.S. government agency securities | 193,302 | 6 | 0 | 193,308 | 0 | 193,308 | 0 | |||||||||||||||||||||||||||||||||||||
| Total financial assets | $ | 383,625 | $ | 6 | $ | (15) | $ | 383,616 | $ | 190,008 | $ | 193,608 | $ | 0 | ||||||||||||||||||||||||||||||
| Financial liabilities: | ||||||||||||||||||||||||||||||||||||||||||||
| Contingent consideration | $ | 11,300 | $ | 0 | $ | 0 | $ | 11,300 | ||||||||||||||||||||||||||||||||||||
| Total financial liabilities | $ | 11,300 | $ | 0 | $ | 0 | $ | 11,300 | ||||||||||||||||||||||||||||||||||||
| December 31, 2019 | ||||||||||||||||||||||||||||||||||||||||||||
| Reported as: | ||||||||||||||||||||||||||||||||||||||||||||
| Cash equivalents | $ | 136,997 | ||||||||||||||||||||||||||||||||||||||||||
| Restricted cash | 6,183 | |||||||||||||||||||||||||||||||||||||||||||
| Marketable securities | 240,436 | |||||||||||||||||||||||||||||||||||||||||||
| Total cash equivalents, restricted cash, and marketable securities | $ | 383,616 | ||||||||||||||||||||||||||||||||||||||||||
| Accrued liabilities | $ | 3,300 | ||||||||||||||||||||||||||||||||||||||||||
| Other long-term liabilities | $ | 8,000 | ||||||||||||||||||||||||||||||||||||||||||
There were 0 transfers between Level 1, Level 2 and Level 3 during the periods presented. The total fair value of investments with unrealized losses at December 31, 2020 was $109.3 million. None of the available-for-sale securities held as of December 31, 2020 have been in an unrealized loss position for more than one year. At December 31, 2020, the remaining contractual maturities of available-for-sale securities ranged from one to nine months. Interest income generated from our investments was $4.0 million and $5.2 million during the years ended December 31, 2020 and 2019, respectively.
Our certificates of deposit and debt securities of U.S. government agency entities are classified as Level 2 as they are valued based upon quoted market prices for similar instruments in active markets, quoted prices for identical or similar instruments in markets that are not active and model-based valuation techniques for which all significant inputs are observable in the market or can be corroborated by observable market data for substantially the full term of the assets. Where applicable, these models project future cash flows and discount the future amounts to a present value using market-based observable inputs obtained from various third-party data providers, including but not limited to benchmark yields, interest rate curves, reported trades, broker/dealer quotes and reference data.
Stock payable liabilities relate to certain indemnification hold-backs resulting from business combinations that are settled in shares of our common stock. We elected to account for these liabilities using the fair value option due to the inherent nature of the liabilities and the changes in value of the underlying shares that will ultimately be issued to settle the liabilities. The estimated fair value of these liabilities is classified as Level 3 and determined based upon the number of shares that are issuable to the sellers and the quoted closing price of our common stock as of the reporting date. The number of shares that will ultimately be issued is subject to adjustment for indemnified claims that existed as of the closing date for each acquisition. Changes in the number of shares issued and share price can significantly affect the estimated fair value of the liabilities. During the year ended December 31, 2020, the change in fair value related to stock payable liabilities recorded to other income (expense), net was expense of $37.5 million.
107
8. Commitments and contingencies
Leases
In 2015, we entered into an operating lease agreement for our headquarters and main production facility in San Francisco, California which commenced in 2016. This lease expires in 2026 and we may renew the lease for an additional ten years. This optional period was not considered reasonably certain to be exercised and therefore we determined the lease term to be a ten-year period expiring in 2026. In connection with the execution of the lease, we provided a security deposit of approximately $4.6 million which is included in restricted cash in our consolidated balance sheets. We also have other operating leases for office and laboratory space in California, Colorado, Massachusetts, New York and Washington and internationally in Australia and Israel. We expect to enter into new leases and modify existing leases as we support continued growth of our operations.
We have entered into various finance lease agreements to obtain laboratory equipment. The terms of our finance leases are generally three years and are typically secured by the underlying equipment. The portion of the future payments designated as principal repayment and related interest was classified as a finance lease obligation on our consolidated balance sheets.
Supplemental information regarding our operating and finance leases were as follows:
| Year Ended December 31, | ||||||||||||||
| 2020 | 2019 | |||||||||||||
| Weighted-average remaining lease term: | ||||||||||||||
| Operating leases | 5.4 years | 6.5 years | ||||||||||||
| Finance leases | 2.6 years | 2.0 years | ||||||||||||
| Weighted-average discount rate: | ||||||||||||||
| Operating leases | 10.6 | % | 11.8 | % | ||||||||||
| Finance leases | 4.8 | % | 5.5 | % | ||||||||||
| Cash payments included in the measurement of lease liabilities (in millions): | ||||||||||||||
| Operating leases | $ | 11.6 | $ | 10.2 | ||||||||||
| Finance leases | $ | 2.0 | $ | 2.1 | ||||||||||
The components of lease costs, which were included in cost of revenue, research and development, selling and marketing and general and administrative expenses on our consolidated statements of operations, were as follows (in thousands):
| Year Ended December 31, | ||||||||||||||||||||
| 2020 | 2019 | 2018 | ||||||||||||||||||
| Operating lease costs | $ | 11,329 | $ | 10,329 | $ | 9,648 | ||||||||||||||
| Sublease income | 0 | (173) | (156) | |||||||||||||||||
| Finance lease costs | 2,084 | 1,546 | 1,820 | |||||||||||||||||
| Total lease costs | $ | 13,413 | $ | 11,702 | $ | 11,312 | ||||||||||||||
Future payments under operating and finance leases as of December 31, 2020 are as follows (in thousands):
| Operating leases | Finance leases | ||||||||||
| 2021 | $ | 14,338 | $ | 2,006 | |||||||
| 2022 | 13,788 | 1,908 | |||||||||
| 2023 | 13,229 | 1,199 | |||||||||
| 2024 | 13,407 | 26 | |||||||||
| 2025 | 11,672 | 0 | |||||||||
| Thereafter | 9,499 | 0 | |||||||||
| Future non-cancelable minimum lease payments | 75,933 | 5,139 | |||||||||
| Less: interest | (18,787) | (321) | |||||||||
| Total lease liabilities | 57,146 | 4,818 | |||||||||
| Less: current portion | (8,789) | (1,695) | |||||||||
| Lease obligations, net of current portion | $ | 48,357 | $ | 3,123 | |||||||
108
Debt financing
In November 2018, we entered into a Note Purchase Agreement (the "2018 Note Purchase Agreement") pursuant to which we were eligible to borrow an aggregate principal amount up to $200.0 million over a seven year maturity term which included an initial borrowing of $75.0 million in November 2018. We received net proceeds of $10.3 million after terminating and repaying the balance of our obligations of approximately $64.7 million with our previous lender.
In September 2019, we settled our obligations under the 2018 Note Purchase Agreement in full for $85.7 million, which included repayment of principal of $75.0 million, accrued interest of $2.4 million, and prepayment fees of $8.9 million which were recorded as debt extinguishment costs in other expense, net in our consolidated statement of operations during the year ended December 31, 2019.
In October 2020, we entered into a credit agreement with a financial institution under which we borrowed $135.0 million (the "2020 Term Loan") concurrent with the closing of the ArcherDX transaction (the "closing date"). The 2020 Term Loan is secured by a first priority lien on all of our and our subsidiaries' assets (including our intellectual property), and is guaranteed by our subsidiaries, in each case, excluding certain excluded assets and immaterial foreign subsidiaries. The 2020 Term Loan bears interest at an annual rate equal to LIBOR, subject to a 2.00% LIBOR floor, plus a margin of 8.75%. The 2020 Term Loan will mature on (i) June 1, 2024 if at such time our Convertible Senior Notes are outstanding and are due to mature on September 1, 2024 (provided that if, prior to such date, the maturity date of at least 80% of the Convertible Senior Notes is extended to a date that is prior to September 1, 2025 the maturity date for the 2020 Term Loan will be automatically extended to a date that is 90 days prior to such Convertible Senior Notes maturity date as extended), or (ii) otherwise, on June 1, 2025. The full amount of the 2020 Term Loan is due upon maturity. If the 2020 Term Loan is prepaid (whether such prepayment is optional or mandatory), we must pay a prepayment fee of 6% if the prepayment occurs prior to the third anniversary of the closing date or 4% if the prepayment occurs after the third anniversary of the closing date and we must also pay a make-whole fee if the prepayment occurs prior to the second anniversary of the closing date. In connection with the 2020 Term Loan, we issued warrants to purchase 1.0 million shares of our common stock with an exercise price of $16.85 per share, exercisable through October 2027. The warrants, which were classified as equity, were recorded at an amount based on the allocated proceeds and do not require subsequent remeasurement. In October 2020, these warrants were exercised in full through net settlement resulting in the issuance of 0.7 million shares.
The credit agreement contains customary events of default and covenants, including among others, covenants limiting our ability to incur debt, incur liens, undergo a change in control, merge with or acquire other entities, make investments, pay dividends or other distributions to holders of our equity securities, repurchase stock, and dispose of assets, in each case subject to certain customary exceptions. In addition, the credit agreement contains financial covenants that require us to maintain a minimum cash balance and minimum quarterly revenue levels.
Debt discounts, including debt issuance costs, related to the 2020 Term Loan of $32.8 million were recorded as a direct deduction from the debt liability and are being amortized to interest expense over the term of the 2020 Term Loan. Interest expense related to our debt financings, excluding the impact of our Convertible Senior Notes, was $7.4 million, $5.7 million and $6.7 million for the years ended December 31, 2020, 2019 and 2018, respectively.
Convertible Senior Notes
In September 2019, we issued, at par value, $350.0 million aggregate principal amount of 2.00% Convertible Senior Notes due 2024 in a private offering. The Convertible Senior Notes are our senior unsecured obligations and will mature on September 1, 2024, unless earlier converted, redeemed or repurchased. The Convertible Senior Notes bear cash interest at a rate of 2.0% per year, payable semi-annually in arrears on March 1 and September 1 of each year, beginning on March 1, 2020.
Upon conversion, the Convertible Senior Notes will be convertible into cash, shares of our common stock or a combination of cash and shares of our common stock, at our election. Our current intent is to settle the principal amount of the Convertible Senior Notes in cash upon conversion, with any remaining conversion value being delivered in shares of our common stock. The initial conversion rate for the Convertible Senior Notes is 33.6293 shares of our common stock per $1,000 principal amount of the Convertible Senior Notes (equivalent to an initial conversion price of approximately $29.74 per share of common stock).
If we undergo a fundamental change (as defined in the indenture governing the Convertible Senior Notes), the holders of the Convertible Senior Notes may require us to repurchase all or any portion of their Convertible Senior Notes for cash at a repurchase price equal to 100% of the principal amount of the Convertible Senior Notes to be repurchased plus accrued and unpaid interest to, but excluding, the redemption date.
109
The Convertible Senior Notes will be convertible at the option of the holders at any time prior to the close of business on the business day immediately preceding March 1, 2024, only under the following circumstances: (1) during any calendar quarter commencing after the calendar quarter ending on December 31, 2019 (and only during such calendar quarter), if the last reported sale price of our common stock for at least 20 trading days (whether or not consecutive) during a period of 30 consecutive trading days ending on, and including, the last trading day of the immediately preceding calendar quarter is greater than or equal to 130% of the conversion price for the Convertible Senior Notes on each applicable trading day; (2) during the 5 business day period after any five consecutive trading day period (the “measurement period”) in which the trading price per $1,000 principal amount of Convertible Senior Notes for each trading day of the measurement period was less than 98% of the product of the last reported sale price of our common stock and the conversion rate on each such trading day; (3) if we call any or all of the Convertible Senior Notes for redemption, at any time prior to the close of business on the scheduled trading day immediately preceding the redemption date; or (4) upon the occurrence of specified corporate events. On or after March 1, 2024 until the close of business on the business day immediately preceding the maturity date, holders may convert their Convertible Senior Notes at any time, regardless of the foregoing circumstances. As of December 31, 2020, none of the above circumstances had occurred and therefore the Convertible Senior Notes could not have been converted. However, these notes were convertible at the option of the holders during the quarter beginning on January 1, 2021 due to the sale price of our common stock during the quarter ended December 31, 2020.
We may not redeem the Convertible Senior Notes prior to September 6, 2022. We may redeem for cash all or any portion of the Convertible Senior Notes, at our option, on or after September 6, 2022 and on or before the 30th scheduled trading day immediately before the maturity date if the last reported sale price of the Common Stock has been at least 130% of the conversion price then in effect for at least 20 trading days (whether or not consecutive) during any 30 consecutive trading day period (including the last trading day of such period) ending on, and including, the trading day immediately preceding the date on which we provide notice of redemption at a redemption price equal to 100% of the principal amount of the notes to be redeemed, plus accrued and unpaid interest to, but excluding, the redemption date.
The Convertible Senior Notes consisted of the following (in thousands):
| December 31, | |||||||||||
| 2020 | 2019 | ||||||||||
| Outstanding principal | $ | 350,000 | $ | 350,000 | |||||||
| Unamortized debt discount and issuance costs | (66,276) | (81,245) | |||||||||
| Net carrying amount, liability component | $ | 283,724 | $ | 268,755 | |||||||
As of December 31, 2020, the fair value of the Convertible Senior Notes was $586.0 million. The estimated fair value of the Convertible Senior Notes, which are classified as Level 2 financial instruments, was determined based on the estimated or actual bid prices of the Convertible Senior Notes in an over-the-counter market. We recognized $22.0 million and $6.5 million of interest expense related to the Convertible Senior Notes during the years ended December 31, 2020 and 2019, respectively.
Guarantees and indemnifications
As permitted under Delaware law and in accordance with our bylaws, we indemnify our directors and officers for certain events or occurrences while the officer or director is or was serving in such capacity. The maximum amount of potential future indemnification is unlimited; however, we maintain director and officer liability insurance. This insurance allows the transfer of the risk associated with our exposure and may enable us to recover a portion of any future amounts paid. We believe the fair value of these indemnification agreements is minimal. Accordingly, we did not record any liabilities associated with these indemnification agreements at December 31, 2020 or 2019.
110
Other commitments
In the normal course of business, we enter into various purchase commitments primarily related to service agreements and laboratory supplies. At December 31, 2020, our total future payments under noncancelable unconditional purchase commitments having a remaining term of over one year were as follows (in thousands):
| Amount | |||||
| 2021 | 23,064 | ||||
| 2022 | 20,372 | ||||
| 2023 | 19,451 | ||||
| 2024 | 9,220 | ||||
| 2025 | 4,530 | ||||
| Thereafter | 25,501 | ||||
| Total | $ | 102,138 | |||
In December 2020, we entered into a lease agreement in San Francisco, California for additional office and lab space. We determined the lease commencement date to be in January 2021 when we took possession of the leased premises. Total lease payments over the course of this lease will be $45.0 million and are included in the purchase commitments above.
Contingencies
We were not a party to any material legal proceedings at December 31, 2020, or at the date of this report except for matters listed below which are related to ArcherDX which we acquired in October 2020. We cannot currently predict the outcome of these actions. The outcome of any such proceedings, regardless of the merits, is inherently uncertain. If we were unable to prevail in any such proceedings, our consolidated financial position, results of operations, and future cash flows may be materially impacted. In addition, we are and may from time to time become involved in various legal proceedings and claims arising in the ordinary course of business. While we believe any such claims are unsubstantiated, and we believe we are in compliance with applicable laws and regulations applicable to our business, the resolution of any such claims could be material.
111
Natera, Inc.
On January 27, 2020, Natera filed a lawsuit against ArcherDX (a subsidiary of Invitae effective October 2, 2020) in the United States District Court for the District of Delaware, alleging that ArcherDX’s products using AMP chemistry, and the manufacture, use, sale, and offer for sale of such products, infringe U.S. Patent No. 10,538,814. On March 25, 2020, ArcherDX filed an answer denying Natera’s allegations and asserting certain affirmative defenses and counterclaims, including that U.S. Patent No. 10,538,814 is invalid and not infringed. On April 15, 2020, Natera filed an answer denying ArcherDX’s counterclaims and filed an amended complaint alleging that ArcherDX’s products using AMP chemistry, including STRATAFIDE, PCM, LiquidPlex, ArcherMET, FusionPlex, and VariantPlex, and the manufacture, use, sale, and offer for sale of such products, infringe U.S. Patent No. 10,538,814, U.S. Patent No. 10,557,172, U.S. Patent No. 10,590,482, and U.S. Patent No. 10,597,708, each of which are held by Natera. Natera seeks, among other things, damages and other monetary relief, costs and attorneys’ fees, and an order enjoining ArcherDX from further infringement of such patents. On May 13, 2020, ArcherDX filed an answer to Natera’s amended complaint denying Natera’s allegations and asserting certain affirmative defenses and counterclaims, including that the asserted patents are invalid and not infringed. On June 3, 2020, Natera filed an answer denying ArcherDX’s counterclaims. On June 4, 2020, ArcherDX filed a motion seeking dismissal of Natera’s infringement claims against STRATAFIDE, PCM, and ArcherMET, and for a judgment that U.S. Patent No. 10,538,814, U.S. Patent No. 10,557,172, and U.S. Patent No. 10,590,482 are invalid. On August 6, 2020, Natera filed another complaint against ArcherDX in the United States District Court for the District of Delaware alleging that ArcherDX’s products using AMP chemistry, including STRATAFIDE, PCM, LiquidPlex, ArcherMET, and VariantPlex, and the manufacture, use, sale, and offer for sale of such products, infringe U.S. Patent No. 10,731,220. Natera seeks, among other things, damages and other monetary relief, costs and attorneys’ fees, and an order enjoining ArcherDX from further infringement of the patent. On October 13, 2020, the court issued an order denying ArcherDX's motion for dismissal of Natera’s infringement claims against STRATAFIDE, PCM, and ArcherMET, and declined to enter judgment that U.S. Patent No. 10,538,814, U.S. Patent No. 10,557,172, and U.S. Patent No. 10,590,482 are invalid. On January 12, 2021, the court issued an order granting Natera leave to amend its complaint to add Invitae as a co-defendant and plead allegations that ArcherDX and Invitae induce end-users to infringe the patents-in-suit. Natera filed its Second Amended Complaint on the same day, with service completed on January 15, 2021. ArcherDX and Invitae filed answers to the Second Amended Complaint on January 26, 2021 and February 5, 2021, respectively, denying Natera's allegations and restating certain affirmative defenses and counterclaims of non-infringement and invalidity. The litigations have now been consolidated for all purposes, are ongoing, and trial has been scheduled for May 2022.
QIAGEN Sciences
On July 10, 2018, ArcherDX and the General Hospital Corporation d/b/a Massachusetts General Hospital, which we refer to as MGH, filed a lawsuit in the United States District Court for the District of Delaware against QIAGEN Sciences, LLC, QIAGEN LLC, QIAGEN Beverly, Inc., QIAGEN Gaithersburg, Inc., QIAGEN GmbH and QIAGEN N.V., which is collectively referred to herein as QIAGEN, and a named QIAGEN executive who was a former member of ArcherDX’s board of directors, alleging several causes of action, including infringement of the ’810 Patent, trade secret misappropriation, breach of fiduciary duty, false advertising, tortious interference and deceptive trade practices. The ’810 Patent relates to methods for preparing a nucleic acid for sequencing and aspects of ArcherDX’s AMP technology. On October 30, 2019, with the permission of the Court, ArcherDX amended ArcherDX’s complaint to add a claim for infringement of the ’597 Patent. The ’597 Patent relates to methods of preparing and analyzing nucleic acids, such as by enriching target sequences prior to sequencing, and aspects of ArcherDX’s AMP technology. The QIAGEN products that ArcherDX alleges infringe the ’810 Patent and the ’597 Patent include, but are not limited to, QIAseq Targeted DNA Panels, QIAseq Targeted RNAscan Panels, QIAseq Index Kits and QIAseq Immune Repertoire RNA Library Kits. ArcherDX is seeking, among other things, damages for ArcherDX’s lost profits due to QIAGEN’s infringement and a permanent injunction enjoining QIAGEN from marketing and selling the infringing products and from using ArcherDX’s trade secrets. On December 5, 2019, QIAGEN and the named QIAGEN executive submitted their answer denying the allegations in ArcherDX’s complaint and asserting affirmative defenses that, among other things, the ’810 Patent and ’597 Patent are not infringed by QIAGEN’s products, that both patents are invalid, and that the complaint fails to state any claim for which relief may be granted. This litigation is ongoing, and trial is currently scheduled for August 2021.
112
9. Stockholders’ equity
Shares outstanding
Shares of convertible preferred and common stock were as follows (in thousands):
| Year Ended December 31, | ||||||||||||||||||||
| 2020 | 2019 | 2018 | ||||||||||||||||||
| Convertible preferred stock: | ||||||||||||||||||||
| Shares outstanding, beginning of period | 125 | 3,459 | 3,459 | |||||||||||||||||
| Conversion into common stock | 0 | (3,334) | 0 | |||||||||||||||||
| Shares outstanding, end of period | 125 | 125 | 3,459 | |||||||||||||||||
| Common stock: | ||||||||||||||||||||
| Shares outstanding, beginning of period | 98,796 | 75,481 | 53,597 | |||||||||||||||||
| Common stock issued in private placement | 16,320 | 0 | 374 | |||||||||||||||||
| Common stock issued in connection with public offering | 24,005 | 11,136 | 17,103 | |||||||||||||||||
| Common stock issued on exercise of stock options, net | 2,659 | 468 | 351 | |||||||||||||||||
| Common stock issued pursuant to vesting of RSUs | 5,304 | 2,683 | 1,369 | |||||||||||||||||
| Common stock issued pursuant to exercises of warrants | 968 | 31 | 1,099 | |||||||||||||||||
| Common stock issued pursuant to employee stock purchase plan | 671 | 455 | 566 | |||||||||||||||||
| Common stock issued pursuant to acquisitions | 37,163 | 5,208 | 1,022 | |||||||||||||||||
| Common stock issued upon conversion of preferred stock | 0 | 3,334 | 0 | |||||||||||||||||
| Shares outstanding, end of period | 185,886 | 98,796 | 75,481 | |||||||||||||||||
2018 Sales Agreement
In August 2018, we entered into a Common Stock Sales Agreement (the “2018 Sales Agreement”) with Cowen and Company, LLC (“Cowen”), under which we could offer and sell from time to time at our sole discretion shares of our common stock through Cowen as our sales agent, in an aggregate amount not to exceed $75.0 million. Cowen may sell the shares by any method permitted by law deemed to be an “at the market” offering as defined in Rule 415 of the Securities Act of 1933, including without limitation sales made directly on The New York Stock Exchange, and also may sell the shares in privately negotiated transactions, subject to our prior approval. Per the terms of the agreement, Cowen receives a commission equal to 3% of the gross proceeds of the sales price of all shares sold through it as sales agent under the 2018 Sales Agreement. In March 2019, we amended the 2018 Sales Agreement to increase the aggregate amount of our common stock to be sold under this agreement to an amount not to exceed $175.0 million.
During the year ended December 31, 2020, we sold a total of 3.6 million shares of common stock under the 2018 Sales Agreement at an average price of $26.33 per share, for gross proceeds of $93.7 million and net proceeds of $90.7 million.
During the year ended December 31, 2019, we sold a total of 0.8 million shares of common stock under the 2018 Sales Agreement at an average price of $25.71 per share, for gross proceeds of $20.2 million and net proceeds of $19.5 million.
During the year ended December 31, 2018, we sold a total of 4.3 million shares of common stock under the 2018 Sales Agreement at an average price of $14.13 per share, for aggregate gross proceeds of $61.1 million and net proceeds of $58.9 million.
Public offerings
In January 2021, we issued, in an underwritten public offering, an aggregate of 8.9 million shares of our common stock at a price of $51.50 per share, for gross proceeds of $460.0 million and net proceeds of approximately $434.3 million.
In April 2020, we issued, in an underwritten public offering, an aggregate of 20.4 million shares of our common stock at a price of $9.00 per share, for gross proceeds of $184.0 million and net proceeds of $173.0 million.
113
In March 2019, we sold, in an underwritten public offering, an aggregate of 10.4 million shares of our common stock at a price of $19.00 per share, for gross proceeds of $196.7 million and net proceeds of $184.5 million.
Private placement
In August 2017, in a private placement to certain accredited investors, we issued 5.2 million shares of common stock at a price of $8.50 per share, and 3.5 million shares of our Series A convertible preferred stock at a price of $8.50 per share, for gross proceeds of approximately $73.5 million and net proceeds of $68.9 million. The Series A preferred stock is convertible into common stock on a 1-for-one basis, subject to adjustment for events such as stock splits, combinations and the like. During the year ended December 31, 2019, 3.3 million shares of Series A convertible preferred stock were converted to 3.3 million shares of common stock.
In connection with our acquisition of ArcherDX, in June 2020 we entered into a definitive agreement to sell $275.0 million in common stock in a private placement at a price of $16.85 per share. The private placement closed in October 2020, concurrently with our acquisition of ArcherDX. We received proceeds of $5.0 million from the private placement during September 2020 and the remainder of the proceeds were received in October 2020.
Common stock warrants
As of December 31, 2020, we had outstanding warrants to purchase common stock as follows:
| Warrant | Issuance Date | Expiration Date | Exercise Price Per Share | Number of Shares of Common Stock Underlying Warrants | ||||||||||||||||||||||
| Warrants issued in exchange for CombiMatrix Series F warrants | November 2017 | March 2021 | $ | 5.95 | 214,154 | |||||||||||||||||||||
The exercise price of warrants issued in exchange for CombiMatrix Series F warrants was determined pursuant to the terms of the acquisition.
10. Stock incentive plans
Stock incentive plans
In 2010, we adopted the 2010 Incentive Plan (the “2010 Plan”). The 2010 Plan provides for the granting of stock-based awards to employees, directors and consultants under terms and provisions established by our Board of Directors. Under the terms of the 2010 Plan, options may be granted at an exercise price not less than the fair market value of our common stock. For employees holding more than 10% of the voting rights of all classes of stock, the exercise prices for incentive and nonstatutory stock options must be at least 110% of fair market of our common stock on the grant date, as determined by our Board of Directors. The terms of options granted under the 2010 Plan may not exceed ten years.
In January 2015, we adopted the 2015 Stock Incentive Plan (the “2015 Plan”), which became effective upon the closing of our initial public offering (“IPO”). Shares outstanding under the 2010 Plan were transferred to the 2015 Plan upon effectiveness of the 2015 Plan. The 2015 Plan provides for automatic annual increases in shares available for grant, beginning on January 1, 2016 through January 1, 2025. In addition, shares subject to awards under the 2010 Plan that are forfeited or terminated will be added to the 2015 Plan. The 2015 Plan provides for the grant of incentive stock options, nonstatutory stock options, restricted stock awards, stock units, stock appreciation rights and other forms of equity compensation, all of which may be granted to employees, including officers, non-employee directors and consultants. Additionally, the 2015 Plan provides for the grant of cash-based awards.
Options granted generally vest over a period of four years. Typically, the vesting schedule for options granted to newly hired employees provides that 1/4 of the award vests upon the first anniversary of the employee’s date of hire, with the remainder of the award vesting monthly thereafter at a rate of 1/48 of the total shares subject to the option. All other options typically vest in equal monthly installments over the four-year vesting schedule. Upon the acquisition of ArcherDX, any option that was outstanding was converted into a fully vested option to purchase a share of our common stock which resulted in the issuance of 3.7 million options.
RSUs generally vest over a period of three years. Typically, the vesting schedule for RSUs provides that 1/3 of the award vests upon each anniversary of the grant date, with certain awards that include a portion that vests immediately upon grant. In June 2019, we granted Time-based RSUs in connection with the acquisition of Singular Bio which vest in three equal installments over a period of 18 months and PRSUs that vest based on the achievement of performance conditions. In December 2020, we granted RSUs in connection with an asset acquisition which vest in two equal installments in December 2021 and December 2022, subject to the employee's continued service with us.
114
Under our management incentive compensation plan, in July 2019 we granted PRSUs to our executive officers as well as other specified senior level employees based on the level of achievement of a specified 2019 revenue goal. One-third of the 0.8 million shares that were ultimately awarded under this plan vested during the year ended December 31, 2020 and the remaining shares will vest through March 2022. In June 2020, we granted 0.3 million PRSUs under this plan which are based on the level of achievement of a specified 2020 cash burn goal. Upon achievement of the specified 2020 cash burn goal, 0.3 million shares were ultimately awarded and began vesting in 2021 over a one year period. These PRSUs had a grant date fair value of $4.2 million based on an estimated issuance of 0.3 million shares and expectation of the performance conditions. During the year ended December 31, 2020, $2.1 million was recorded as stock-based compensation expense related to the awards. NaN PRSUs were granted during the year ended December 31, 2018.
Activity under the 2010 Plan and the 2015 Plan is set forth below (in thousands, except per share amounts and years):
| Shares Available For Grant | Stock Options Outstanding | Weighted-Average Exercise Price Per Share | Weighted-Average Remaining Contractual Life (years) | Aggregate Intrinsic Value | |||||||||||||||||||||||||
| Balance at December 31, 2019 | 5,444 | 3,542 | $ | 9.49 | 6.1 | $ | 24,966 | ||||||||||||||||||||||
| Additional shares reserved | 9,019 | — | |||||||||||||||||||||||||||
| Options granted | (4,005) | 4,005 | $ | 3.74 | |||||||||||||||||||||||||
| Options cancelled | 11 | (11) | $ | 7.05 | |||||||||||||||||||||||||
| Options exercised | — | (2,659) | $ | 4.04 | |||||||||||||||||||||||||
RSUs and PRSUs granted(1) | (3,502) | — | |||||||||||||||||||||||||||
| RSUs and PRSUs cancelled | 480 | — | |||||||||||||||||||||||||||
| Balance at December 31, 2020 | 7,447 | 4,877 | $ | 7.75 | 6.8 | $ | 166,130 | ||||||||||||||||||||||
| Options exercisable at December 31, 2020 | 4,432 | $ | 6.86 | 6.6 | $ | 154,907 | |||||||||||||||||||||||
| Options vested and expected to vest at December 31, 2020 | 4,809 | $ | 7.62 | 6.7 | $ | 164,410 | |||||||||||||||||||||||
(1) Includes the changes in time-based RSUs and PRSUs granted as a part of the Singular Bio acquisition in June 2019 and shares granted in an asset acquisition in December 2020 which are based on a fixed dollar value. The number of shares issued will be variable until the awards vest.
The aggregate intrinsic value is calculated as the difference between the exercise price of the underlying stock options and the fair value of our common stock for stock options that were in-the-money.
The weighted-average fair value of options to purchase common stock granted was $10.10, $14.52 and $4.87 in the years ended December 31, 2020, 2019 and 2018, respectively.
The total grant-date fair value of options to purchase common stock vested was $2.8 million, $4.3 million and $5.9 million in the year ended December 31, 2020, 2019, and 2018, respectively.
The intrinsic value of options to purchase common stock exercised was $104.4 million, $6.3 million and $1.7 million in the years ended December 31, 2020, 2019 and 2018, respectively.
115
The following table summarizes RSU, including PRSU, activity (in thousands, except per share data):
| Number of Shares | Weighted-Average Grant Date Fair Value Per Share | ||||||||||
| Balance at December 31, 2019 | 8,885 | $ | 15.17 | ||||||||
| RSUs granted | 4,874 | $ | 20.35 | ||||||||
Time-based RSUs and PRSUs granted - variable (1) | (1,646) | $ | 24.12 | ||||||||
| PRSUs granted | 274 | $ | 16.17 | ||||||||
| RSUs vested | (5,305) | $ | 19.76 | ||||||||
| RSUs cancelled | (480) | $ | 18.23 | ||||||||
| Balance at December 31, 2020 | 6,602 | $ | 12.89 | ||||||||
(1) Includes the changes in the Time-based RSUs and PRSUs granted as a part of the Singular Bio acquisition in June 2019 and the shares granted in an asset acquisition in December 2020 which are based on a fixed dollar value. The number of shares issued will be variable until the awards vest. The weighted-average grant date fair value per share reflects the fair value pricing of the full award.
2015 Employee Stock Purchase Plan
In January 2015, we adopted the 2015 Employee Stock Purchase Plan (the “ESPP”), which became effective upon the closing of the IPO. Employees participating in the ESPP may purchase common stock at 85% of the lesser of the fair market value of common stock on the purchase date or last trading day preceding the offering date. At December 31, 2020, cash received from payroll deductions pursuant to the ESPP was $1.8 million.
The ESPP provides for automatic annual increases in shares available for grant, beginning on January 1, 2016 and continuing through January 1, 2025. At December 31, 2020, a total of 0.9 million shares of common stock are reserved for issuance under the ESPP.
Stock-based compensation
We use the grant date fair value of our common stock to value options when granted. In determining the fair value of stock options and ESPP purchases, we use the Black-Scholes option-pricing model and, for stock options, the assumptions discussed below. Each of these inputs is subjective and its determination generally requires significant judgment. The fair value of RSU and PRSU awards is based on the grant date share price. Compensation cost is recognized as expense on a straight-line basis over the vesting period for options and RSUs and on an accelerated basis for PRSUs.
Expected term—The expected term represents the period that our stock-based awards are expected to be outstanding and is determined using the simplified method (based on the midpoint between the vesting date and the end of the contractual term).
Expected volatility—We estimate expected volatility based on the historical volatility of our common stock over a period equal to the expected term of stock option grants and RSUs and over the expected six-month term ESPP purchase periods.
Risk-free interest rate—The risk-free interest rate is based on the U.S. Treasury zero coupon issues in effect at the time of grant for periods corresponding with the expected term of the option.
Dividend yield—We have never paid dividends on our common stock and have no plans to pay dividends on our common stock. Therefore, we used an expected dividend yield of zero.
The fair value of share-based payments for stock options granted to employees and directors was estimated on the date of grant using the Black-Scholes option-pricing model based on the following assumptions:
| Year Ended December 31, | |||||||||||||||||
| 2020 | 2019 | 2018 | |||||||||||||||
| Expected term (in years) | 6.0 | 6.0 | 6.0 | ||||||||||||||
| Expected volatility | 71.0% | 64.2% | 59.6% | ||||||||||||||
| Risk-free interest rate | 0.5% | 2.6% | 2.8% | ||||||||||||||
The fair value of shares purchased pursuant to the ESPP is estimated using the Black-Scholes option pricing model. For the years ended December 31, 2020, 2019 and 2018, the weighted-average grant date fair value per share for the ESPP was $10.98, $6.05 and $3.26, respectively.
116
The fair value of the shares purchased pursuant to the ESPP was estimated using the following assumptions:
| Year Ended December 31, | |||||||||||||||||
| 2020 | 2019 | 2018 | |||||||||||||||
| Expected term (in years) | 0.5 | 0.5 | 0.5 | ||||||||||||||
| Expected volatility | 105.7% | 66.3% | 71.7% | ||||||||||||||
| Risk-free interest rate | 0.1% | 2.0% | 2.1% | ||||||||||||||
The following table summarizes stock-based compensation expense for the years ended December 31, 2020, 2019 and 2018, included in the consolidated statements of operations (in thousands):
| Year Ended December 31, | |||||||||||||||||
| 2020 | 2019 | 2018 | |||||||||||||||
| Cost of revenue | $ | 8,713 | $ | 4,563 | $ | 2,960 | |||||||||||
| Research and development | 91,762 | 52,450 | 7,017 | ||||||||||||||
| Selling and marketing | 14,418 | 7,641 | 4,887 | ||||||||||||||
| General and administrative | 43,854 | 11,294 | 5,986 | ||||||||||||||
| Total stock-based compensation expense | $ | 158,747 | $ | 75,948 | $ | 20,850 | |||||||||||
At December 31, 2020, unrecognized compensation expense related to unvested stock options, net of estimated forfeitures, was $3.1 million, which we expect to recognize on a straight-line basis over a weighted-average period of 2.4 years. Unrecognized compensation expense related to RSUs, including PRSUs, and awards that are contingently issuable upon the completion of certain milestones related to our acquisition of ArcherDX at December 31, 2020, net of estimated forfeitures, was $144.2 million, which we expect to recognize on a straight-line basis over a weighted-average period of 1.6 years.
11. Income taxes
We recorded a benefit for income taxes in the years ended December 31, 2020, 2019 and 2018. The components of net loss before taxes by U.S. and foreign jurisdictions are as follows (in thousands):
| Year Ended December 31, | |||||||||||||||||
| 2020 | 2019 | 2018 | |||||||||||||||
| United States | $ | 712,409 | $ | 260,531 | $ | 132,194 | |||||||||||
| Foreign | 1,861 | (116) | (39) | ||||||||||||||
| Total | $ | 714,270 | $ | 260,415 | $ | 132,155 | |||||||||||
The components of the provision for income taxes are as follows (in thousands):
| Year Ended December 31, | |||||||||||||||||
| 2020 | 2019 | 2018 | |||||||||||||||
| Current: | |||||||||||||||||
| Foreign | 171 | 85 | 62 | ||||||||||||||
| Total current benefit for income taxes | 171 | 85 | 62 | ||||||||||||||
| Deferred: | |||||||||||||||||
| Federal | (94,279) | (16,011) | (2,862) | ||||||||||||||
| State | (17,730) | (2,524) | 0 | ||||||||||||||
| Foreign | (262) | 0 | 0 | ||||||||||||||
| Total deferred benefit for income taxes | (112,271) | (18,535) | (2,862) | ||||||||||||||
| Total income tax benefit | $ | (112,100) | $ | (18,450) | $ | (2,800) | |||||||||||
117
The following table presents a reconciliation of the tax expense computed at the statutory federal rate and our tax expense for the periods presented:
| Year Ended December 31, | |||||||||||||||||
| 2020 | 2019 | 2018 | |||||||||||||||
| U.S. federal taxes at statutory rate | 21.0 | % | 21.0 | % | 21.0 | % | |||||||||||
| State taxes (net of federal benefit) | 3.4 | % | 3.7 | % | 5.2 | % | |||||||||||
| Stock-based compensation | (1.6) | % | 1.3 | % | (0.7) | % | |||||||||||
| Research and development credits | 1.1 | % | 0 | % | 2.7 | % | |||||||||||
| Non-deductible expenses | (1.5) | % | (1.6) | % | (0.6) | % | |||||||||||
| Change in valuation allowance | (6.7) | % | (17.3) | % | (25.5) | % | |||||||||||
| Total | 15.7 | % | 7.1 | % | 2.1 | % | |||||||||||
The tax effects of temporary differences and carryforwards that give rise to significant portions of the deferred tax assets are as follows (in thousands):
| As of December 31, | |||||||||||
| 2020 | 2019 | ||||||||||
| Deferred tax assets: | |||||||||||
| Net operating loss carryforwards | $ | 337,866 | $ | 173,182 | |||||||
| Tax credits | 19,969 | 0 | |||||||||
| Revenue recognition differences | 9,099 | 5,138 | |||||||||
| Leasing Liabilities | 14,274 | 11,626 | |||||||||
| Accruals and other | 37,677 | 14,391 | |||||||||
| Gross deferred tax assets | 418,885 | 204,337 | |||||||||
| Valuation allowance | (209,308) | (145,318) | |||||||||
| Total deferred tax assets | 209,577 | 59,019 | |||||||||
| Deferred tax liabilities: | |||||||||||
| Amortization and depreciation | (233,150) | (30,875) | |||||||||
| Convertible Senior Notes | (14,658) | (17,720) | |||||||||
| Leasing Assets | (13,307) | (10,424) | |||||||||
| Total deferred tax liabilities | (261,115) | (59,019) | |||||||||
| Net deferred tax liabilities | $ | (51,538) | $ | 0 | |||||||
In December 2017, the Tax Cuts and Jobs Act of 2017 (the “Tax Act”) was signed into law making significant changes to the Internal Revenue Code. Changes included among other items, a reduction of the corporate tax rate from a top marginal rate of 35% to a flat rate of 21%. Although the Tax Act was generally effective January 1, 2018, GAAP required recognition of the tax effects of new legislation during the reporting period that includes the enactment date, which was December 22, 2017. As a result of the lower corporate tax rate enacted as part of the Tax Act, during 2017, the Company recorded a provisional estimate to reduce deferred tax assets by $48.8 million offset by a corresponding reduction in the valuation allowance resulting in no net impact to our income tax benefit or expense.
In December 2017, the Securities and Exchange Commission issued Staff Accounting Bulletin No. 118 ("SAB 118") to address the application of U.S. GAAP in situations when a registrant does not have the necessary information available, prepared, or analyzed (including computations) in reasonable detail to complete the accounting for certain income tax effects of the Tax Act. In accordance with SAB 118, during 2017, we recorded a provisional estimate which resulted in a $48.8 million reduction in deferred tax assets and in the fourth quarter of 2018, we completed our analysis of the impact of the Tax Act and determined that no material adjustments were required to the provisional amounts previously recorded.
We historically established a full valuation allowance against our deferred tax assets due to the uncertainty surrounding realization of such assets. In 2020, we released approximately $112.1 million of our valuation allowance to account for acquired intangibles that support the future realization of some of our deferred tax assets. Due to the overall increase of deferred tax assets, our valuation allowance also increased from the prior year. Our valuation allowance increased by $64.0 million, $23.4 million, and $26.3 million during the years ended December 31, 2020, 2019, and 2018, respectively.
118
As of December 31, 2020, we had net operating loss carryforwards of approximately $1.4 billion and $890.6 million available to reduce future taxable income, if any, for federal and state income tax purposes, respectively. Of the $1.4 billion, $284.9 million will begin to expire in 2030 while $1.1 billion have no expiration date. The state net operating loss carryforwards will begin to expire in 2030.
As of December 31, 2020, we had research and development credit carryforwards of approximately $26.2 million and $17.6 million available to reduce our future tax liability, if any, for federal and state income tax purposes, respectively. The federal credit carryforwards begin to expire in 2030. California credit carryforwards have no expiration date.
Internal Revenue Code ("IRC") section 382 places a limitation (the “Section 382 limitation” or “annual limitation”) on the amount of taxable income that can be offset by net operating loss carryforwards after a change in control (generally greater than 50% change in ownership) of a loss corporation. Similar provisions exist for states. In addition, and as a result of the acquisitions of Good Start Genetics and CombiMatrix in 2017, acquisitions of Singular Bio, Jungla, and Clear Genetics in 2019, and acquisitions of YouScript and ArcherDX in 2020, tax loss carryforwards from acquired entities are also subject to the Section 382 limitation due to the change in control in the acquired entities in the current year.
In addition, as a result of equity issued in connection with various acquisitions, we also performed a section 382 analysis in 2020 with respect to our operating loss and credit carryforwards. We concluded while an ownership change occurred in 2019 as defined under IRC section 382, none of our net operating loss carryforwards would expire unused solely as a result of annual limitations imposed on the use of the carryforwards under IRC sections 382 and 383.
As of December 31, 2020, we had unrecognized tax benefits of $22.0 million, which primarily relates to research and development credits, none of which would currently affect our effective tax rate if recognized due to our valuation allowance against our deferred tax assets. During the year, we benchmarked the reserves of similar tax positions within the industry based on IRS and state audits of comparable companies. Based on our analysis, we decreased our unrecognized tax benefits to more closely align with other comparable companies within the industry. As these reserves relate primarily to research and development credits which have a full valuation allowance, such adjustments did not impact our income tax provision. Unrecognized tax benefits are not expected to materially change in the next 12 months.
A reconciliation of the beginning and ending amount of unrecognized tax benefits is as follows (in thousands):
| Year ended December 31, | |||||||||||||||||
| 2020 | 2019 | 2018 | |||||||||||||||
| Unrecognized tax benefits, beginning of period | $ | 26,985 | $ | 16,375 | $ | 10,561 | |||||||||||
| Gross increases—current period tax positions | 8,368 | 10,311 | 5,686 | ||||||||||||||
| Gross increases—prior period tax positions | 53 | 299 | 128 | ||||||||||||||
| Gross decreases—prior period tax positions | (13,441) | 0 | 0 | ||||||||||||||
| Unrecognized tax benefits, end of period | $ | 21,965 | $ | 26,985 | $ | 16,375 | |||||||||||
Our policy is to include penalties and interest expense related to income taxes as a component of tax expense. We have not accrued interest and penalties related to the unrecognized tax benefits reflected in the financial statements for the years ended December 31, 2020, 2019 and 2018.
Our major tax jurisdictions are the United States and California. All of our tax years will remain open for examination by the Federal and state tax authorities for three and four years, respectively, from the date of utilization of the net operating loss or research and development credit. We do not have any tax audits pending.
12. Net loss per share
The following table presents the calculation of basic and diluted net loss per share (in thousands, except per share data):
| Year ended December 31, | |||||||||||||||||
| 2020 | 2019 | 2018 | |||||||||||||||
| Net loss | $ | (602,170) | $ | (241,965) | $ | (129,355) | |||||||||||
| Shares used in computing net loss per share, basic and diluted | 134,587 | 90,859 | 66,747 | ||||||||||||||
| Net loss per share, basic and diluted | $ | (4.47) | $ | (2.66) | $ | (1.94) | |||||||||||
119
The following common stock equivalents have been excluded from diluted net loss per share because their inclusion would be anti-dilutive (in thousands):
| Year Ended December 31, | |||||||||||||||||
| 2020 | 2019 | 2018 | |||||||||||||||
| Shares of common stock subject to outstanding options | 6,878 | 3,662 | 4,028 | ||||||||||||||
| Shares of common stock subject to outstanding warrants | 405 | 592 | 1,513 | ||||||||||||||
| Shares of common stock subject to outstanding RSUs | 5,590 | 5,293 | 3,476 | ||||||||||||||
| Shares of common stock subject to outstanding PRSUs | 1,658 | 1,860 | 0 | ||||||||||||||
| Shares of common stock pursuant to ESPP | 294 | 239 | 294 | ||||||||||||||
| Shares of common stock underlying Series A convertible preferred stock | 125 | 702 | 3,459 | ||||||||||||||
| Shares of common stock subject to Convertible Senior Notes conversion | 8,371 | 3,612 | 0 | ||||||||||||||
| Total shares of common stock equivalents | 23,321 | 15,960 | 12,770 | ||||||||||||||
13. Geographic information
Revenue by country is determined based on the billing address of the customer and is summarized as follows (in thousands):
| Year Ended December 31, | |||||||||||||||||
| 2020 | 2019 | 2018 | |||||||||||||||
| United States | $ | 255,680 | $ | 202,550 | $ | 138,239 | |||||||||||
| Canada | 4,529 | 4,356 | 4,206 | ||||||||||||||
| Rest of world | 19,389 | 9,918 | 5,254 | ||||||||||||||
| Total revenue | $ | 279,598 | $ | 216,824 | $ | 147,699 | |||||||||||
As of December 31, 2020, 2019 and 2018, our long-lived assets were primarily located in the United States other than operating lease assets representing our right-of-use for leased facilities in Israel and Australia.
14. Selected quarterly data (unaudited)
The following table summarizes our quarterly financial information for 2020 and 2019 (in thousands, except per share amounts):
| Three Months Ended | ||||||||||||||||||||||||||
| March 31, 2020 | June 30, 2020 | September 30, 2020 | December 31, 2020 | |||||||||||||||||||||||
| Revenue | $ | 64,248 | $ | 46,191 | $ | 68,728 | $ | 100,431 | ||||||||||||||||||
| Cost of revenue | $ | 40,422 | $ | 42,952 | $ | 46,643 | $ | 68,258 | ||||||||||||||||||
| Loss from operations | $ | (97,784) | $ | (142,082) | $ | (80,823) | $ | (331,483) | ||||||||||||||||||
| Net loss | $ | (98,527) | $ | (166,403) | $ | (102,902) | $ | (234,338) | ||||||||||||||||||
Net loss per share, basic and diluted(1) | $ | (0.99) | $ | (1.29) | $ | (0.78) | $ | (1.30) | ||||||||||||||||||
| Three Months Ended | ||||||||||||||||||||||||||
| March 31, 2019 | June 30, 2019 | September 30, 2019 | December 31, 2019 | |||||||||||||||||||||||
| Revenue | $ | 40,553 | $ | 53,475 | $ | 56,511 | $ | 66,285 | ||||||||||||||||||
| Cost of revenue | $ | 21,254 | $ | 28,006 | $ | 32,120 | $ | 36,723 | ||||||||||||||||||
| Loss from operations | $ | (36,207) | $ | (51,886) | $ | (76,983) | $ | (79,036) | ||||||||||||||||||
| Net loss | $ | (37,677) | $ | (48,676) | $ | (78,707) | $ | (76,905) | ||||||||||||||||||
Net loss per share, basic and diluted(1) | $ | (0.47) | $ | (0.54) | $ | (0.82) | $ | (0.79) | ||||||||||||||||||
___________________________________________________________________
(1) Net loss per share is computed independently for each of the quarters presented. Therefore, the sum of quarterly net loss per share information may not equal annual net loss per share.
120
15. Subsequent event
In February 2021, we acquired 100% of the equity interest of Reference Genomics, Inc. d/b/a One Codex "One Codex", a company developing and commercializing products and services relating to microbiome sequencing, analysis and reporting, for upfront consideration consisting of 1.2 million shares of our common stock and $17.0 million in cash. Up to approximately $0.1 million in cash and 0.2 million additional shares of our common stock are subject to a hold back to satisfy indemnification obligations that may arise following the closing. Given the timing of the closing of the transaction with One Codex, we are currently in the process of valuing the assets acquired and liabilities assumed. As a result, we are not yet able to provide the amounts to be recognized as of the acquisition date for the major classes of assets acquired and liabilities assumed and other related disclosures. We will disclose this and other related information in our Quarterly Report on Form 10-Q for the three months ending March 31, 2021.
ITEM 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
Not applicable.
ITEM 9A. Controls and Procedures.
Evaluation of disclosure controls and procedures
We maintain “disclosure controls and procedures,” as such term is defined in Rule 13a-15(e) under the Securities Exchange Act of 1934, or Exchange Act, that are designed to ensure that information required to be disclosed by us in reports that we file or submit under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in Securities and Exchange Commission rules and forms, and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial and accounting officer, as appropriate, to allow timely decisions regarding required disclosure. In designing and evaluating our disclosure controls and procedures, management recognized that disclosure controls and procedures, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the disclosure controls and procedures are met. Our disclosure controls and procedures have been designed to meet reasonable assurance standards. Additionally, in designing disclosure controls and procedures, our management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible disclosure controls and procedures. The design of any disclosure controls and procedures also is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions.
Based on their evaluation as of the end of the period covered by this Annual Report on Form 10-K, our Chief Executive Officer (our principal executive officer) and Chief Financial Officer (our principal financial officer) have concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
Changes in internal control over financial reporting
There was no change in our internal control over financial reporting (as defined in Rule 13a-15(f) under the Exchange Act) identified in connection with the evaluation described in Item 9A above that occurred during our last fiscal quarter that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Management’s annual report on internal control over financial reporting
Our management is responsible for establishing and maintaining internal control over our financial reporting. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Because of inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues, if any, within an organization have been detected. Projections of any evaluation of the effectiveness of internal control to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with policies or procedures may deteriorate.
The scope of management’s assessment of the effectiveness of our internal control over financial reporting excludes the operations of ArcherDX, which we acquired in October 2020. This exclusion is in accordance with the SEC’s general guidance that an assessment of a recently acquired business may be omitted from the scope of our evaluation in the year of acquisition. ArcherDX constituted 3% of our consolidated total assets and 6% of our consolidated revenue as of and for the year ended December 31, 2020.
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, assessed the effectiveness of our internal control over financial reporting as of December 31, 2020. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission, or COSO, in Internal Control—Integrated Framework (2013 Framework). Based on the assessment using those criteria, our management concluded that, as of December 31, 2020, our internal control over financial reporting was effective. Our independent registered public accounting firm, Ernst & Young LLP, has issued an audit report with respect to our internal control over financial reporting, which appears in Part II, Item 8 of this Annual Report on Form 10-K.
122
Report of Independent Registered Public Accounting Firm
To the Stockholders and Board of Directors of Invitae Corporation
Opinion on Internal Control Over Financial Reporting
We have audited Invitae Corporation’s internal control over financial reporting as of December 31, 2020, based on criteria established in Internal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). In our opinion, Invitae Corporation (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2020, based on the COSO criteria.
As indicated in the accompanying Management’s Report on Internal Control over Financial Reporting, management’s assessment of and conclusion on the effectiveness of internal control over financial reporting did not include the internal controls of ArcherDX, Inc., which is included in the 2020 consolidated financial statements of the Company and constituted 3% and 1% of total and net assets, respectively, as of December 31, 2020 and 6% and 4% of revenues and net loss, respectively, for the year then ended. Our audit of internal control over financial reporting of the Company also did not include an evaluation of the internal control over financial reporting of ArcherDX, Inc.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated balance sheets of Invitae Corporation as of December 31, 2020 and 2019, the related consolidated statements of operations, comprehensive loss, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2020, and the related notes (collectively referred to as the “consolidated financial statements”) and our report dated February 26, 2021 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
123
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Ernst & Young LLP
Redwood City, California
February 26, 2021
ITEM 9B. Other Information.
None.
124
PART III
ITEM 10. Directors, Executive Officers and Corporate Governance.
The information required by this item with respect to directors is incorporated by reference from the information under the caption “Election of Directors,” contained in our proxy statement to be filed with the Securities and Exchange Commission no later than 120 days from the end of our fiscal year ended December 31, 2020 in connection with the solicitation of proxies for our 2021 Annual Meeting of Stockholders, or the Proxy Statement. Certain information required by this item concerning executive officers is set forth in Part I of this Report under the caption “Information About our Executive Officers” and is incorporated herein by reference.
There have been no material changes to the procedures by which stockholders may recommend nominees to our Board of Directors.
Item 405 of Regulation S-K calls for disclosure of any known late filing or failure by an insider to file a report required by Section 16(a) of the Exchange Act. To the extent disclosure for delinquent reports is being made, it can be found under the caption “Delinquent Section 16(a) Reports” in the Proxy Statement and is incorporated herein by reference.
Our board of directors has adopted a code of business conduct and a code of ethics for senior financial officers applicable to our Chief Executive Officer and Chief Financial Officer as well as other key management employees addressing ethical issues. The code of business conduct and the code of ethics are each posted on our website www.invitae.com. The code of business conduct and the code of ethics can only be amended by the approval of a majority of our board of directors. Any waiver to the code of business conduct for an executive officer or director or any waiver of the code of ethics may only be granted by our board of directors or our nominating and corporate governance committee and must be timely disclosed as required by applicable law. We have implemented whistleblower procedures that establish formal protocols for receiving and handling complaints from employees. Any concerns regarding accounting or auditing matters reported under these procedures will be communicated promptly to our audit committee. Stockholders may request a free copy of our code of business conduct and code of ethics by contacting Invitae Corporation, Attention: Chief Financial Officer, 1400 16th Street, San Francisco, California 94103. None of the materials on, or accessible through, our website is part of this report or incorporated by reference herein.
To date, there have been no waivers under our code of business conduct or code of ethics. We intend to disclose future amendments to certain provisions of our code of business conduct or code of ethics or waivers of such codes granted to executive officers and directors on our website at http://www.invitae.com within four business days following the date of such amendment or waiver.
Our Board of Directors has appointed an Audit Committee, comprised of Geoffrey S. Crouse, Christine M. Gorjanc, and Kimber D. Lockhart. The Board of Directors has determined that each of the members of our Audit Committee qualifies as an Audit Committee Financial Expert under the definition outlined by the Securities and Exchange Commission. In addition, each of the members of the Audit Committee qualifies as an "independent director" under the current rules of the New York Stock Exchange and Securities and Exchange Commission rules and regulations.
ITEM 11. Executive Compensation.
The information required by this item is incorporated by reference from the information under the captions “Election of Directors-Director Compensation” and “Executive Compensation” contained in the Proxy Statement.
ITEM 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The information required by this item is incorporated by reference to the disclosure appearing under the headings “Security Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information” contained in the Proxy Statement.
ITEM 13. Certain Relationships and Related Transactions, and Director Independence.
The information required by this item is incorporated by reference from the information under the captions “Certain Relationships and Related Transactions,” "Corporate Governance" and “Director Independence” contained in the Proxy Statement.
125
ITEM 14. Principal Accountant Fees and Services.
The information required by this item is incorporated by reference from the information under the caption “Ratification of the Appointment of Independent Registered Public Accounting Firm” contained in the Proxy Statement.
126
PART IV
ITEM 15. Exhibits and Financial Statement Schedules.
(a)Documents filed as part of this report
1.Financial Statements: Reference is made to the Index to Financial Statements of Invitae Corporation included in Item 8 of Part II hereof.
2.Financial Statement Schedules: All schedules have been omitted because they are not required, not applicable, or the required information is included in the financial statements or notes thereto.
3.Exhibits: See Item 15(b) below. Each management contract or compensating plan or arrangement required to be filed has been identified.
(b)Exhibits
Exhibit Number | Description | |||||||
2.1@ | ||||||||
2.2@ | ||||||||
2.3@^ | Agreement and Plan of Merger, dated as of November 8, 2019, by and among Invitae Corporation, Catalina Merger Sub A Inc., Catalina Merger Sub B LLC, Clear Genetics, Inc. and Shareholder Representative Services LLC.(incorporated by reference to Exhibit 2.3 to the Registrant's Annual Report on Form 10-K for the year ended December 31, 2019). | |||||||
2.4@^ | ||||||||
2.5@^ | ||||||||
2.6@^ | ||||||||
2.7@ | ||||||||
| 3.1 | ||||||||
| 3.1.1 | ||||||||
| 3.2 | ||||||||
| 4.1* | ||||||||
| 4.2 | ||||||||
| 4.3 | ||||||||
| 4.4 | ||||||||
| 4.5 | ||||||||
127
Exhibit Number | Description | |||||||
| 4.6 | ||||||||
| 4.7 | ||||||||
| 4.8 | ||||||||
| 4.9 | ||||||||
| 4.10 | ||||||||
| 4.11 | ||||||||
| 4.12 | ||||||||
| 4.13 | ||||||||
| 4.14* | ||||||||
10.1# | ||||||||
10.2# | ||||||||
10.3# | ||||||||
10.4# | ||||||||
10.5#* | ||||||||
10.6# | ||||||||
10.7# | ||||||||
10.8# | ||||||||
10.9# | ||||||||
10.10# | ||||||||
128
Exhibit Number | Description | |||||||
10.11#^ | ||||||||
10.12# | ||||||||
| 10.13 | ||||||||
| 10.14 | ||||||||
| 10.15 | ||||||||
| 10.16 | ||||||||
10.17# | ||||||||
| 10.18 | ||||||||
| 10.19 | ||||||||
10.20^ | ||||||||
10.21#* | ||||||||
| 21.1* | ||||||||
| 23.1* | ||||||||
| 24.1* | Power of Attorney (contained on the signature page to this Form 10-K). | |||||||
| 31.1* | ||||||||
| 31.2* | ||||||||
32.1+ | ||||||||
32.2+ | ||||||||
| 101.INS* | Inline XBRL Instance Document - the instance document does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document. | |||||||
| 101.SCH* | Inline XBRL Taxonomy Extension Schema | |||||||
| 101.CAL* | Inline XBRL Taxonomy Extension Calculation Linkbase | |||||||
| 101.DEF* | Inline XBRL Taxonomy Extension Definition Linkbase | |||||||
| 101.LAB* | Inline XBRL Taxonomy Extension Label Linkbase | |||||||
| 101.PRE* | Inline XBRL Taxonomy Extension Presentation Linkbase | |||||||
| 104 | Cover Page Interactive Data File (the cover page XBRL tags are embedded within the Inline XBRL document and included as Exhibit 101). | |||||||
__________________________________________
129
| # | Indicates management contract or compensatory plan or arrangement. | ||||
| * | Filed herewith. | ||||
| @ | The schedules and exhibits to this agreement have been omitted pursuant to Item 601(b)(2) of Regulation S-K. A copy of any omitted schedule and/or exhibit will be furnished to the SEC upon request. | ||||
| ^ | Portions of this Exhibit have been redacted in accordance with Item 601 of Regulation S-K | ||||
Copies of the above exhibits not contained herein are available to any stockholder, upon payment of a reasonable per page fee, upon written request to: Chief Financial Officer, Invitae Corporation, 1400 16th Street, San Francisco, California 94103.
(c)Financial Statement Schedules: Reference is made to Item 15(a) 2 above.
ITEM 16. Form 10-K Summary.
Not applicable.
130
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| INVITAE CORPORATION | ||||||||
| By: | /s/ Sean E. George, Ph.D. | |||||||
Sean E. George, Ph.D. President and Chief Executive Officer | ||||||||
Date: February 26, 2021
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Sean E. George and Shelly D. Guyer, and each of them, his true and lawful attorneys-in-fact, each with full power of substitution, for him or her in any and all capacities, to sign any amendments to this report on Form 10-K and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that each of said attorneys-in-fact or their substitute or substitutes may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons, on behalf of the registrant on the dates and the capacities indicated.
| Signature | Title | Date | ||||||||||||
| /s/ Sean E. George, Ph.D. | President and Chief Executive Officer (Principal Executive Officer) and Director | February 26, 2021 | ||||||||||||
| Sean E. George, Ph.D. | ||||||||||||||
| /s/ Shelly D. Guyer | Chief Financial Officer (Principal Financial Officer) | February 26, 2021 | ||||||||||||
| Shelly D. Guyer | ||||||||||||||
| /s/ Robert F. Werner | Chief Accounting Officer (Principal Accounting Officer) | February 26, 2021 | ||||||||||||
| Robert F. Werner | ||||||||||||||
| /s/ Eric Aguiar, M.D. | Director | February 26, 2021 | ||||||||||||
| Eric Aguiar, M.D. | ||||||||||||||
| /s/ Geoffrey S. Crouse | Director | February 26, 2021 | ||||||||||||
| Geoffrey S. Crouse | ||||||||||||||
| /s/ Christine M. Gorjanc | Director | February 26, 2021 | ||||||||||||
| Christine M. Gorjanc | ||||||||||||||
| /s/ Kimber D. Lockhart | Director | February 26, 2021 | ||||||||||||
| Kimber D. Lockhart | ||||||||||||||
| /s/ Jason W. Myers | Director | February 26, 2021 | ||||||||||||
| Jason W. Myers | ||||||||||||||
| /s/ Chitra Nayak | Director | February 26, 2021 | ||||||||||||
| Chitra Nayak | ||||||||||||||
131