UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
10-K
(Mark One)
| ☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2021
OR
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number
001-39894

GREENLIGHT BIOSCIENCES HOLDINGS, PBC
(Exact name of registrant as specified in its charter)
Delaware | 200 Boston Avenue Medford, MA 02155 (617) 616-8188 | 85-1914700 | ||
(State or other jurisdiction of incorporation or organization) | (Address, including offices) | (I.R.S. Employer Identification No.) |
Securities registered pursuant to Section 12(b) of the Act:
Title of Each Class | Trading Symbol(s) | Name of Each Exchange on Which Registered | ||
| Common stock, par value $0.0001 per share | GRNA | The Nasdaq Stock Market LLC | ||
Warrants, each exercisable for one share of Common Stock for $11.50 per share | GRNAW | The Nasdaq Stock Market LLC |
Securities registered pursuant to section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act
. Y
es
☐ No☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act
. Y
es
☐ No ☒
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports); and (2) has been subject to such filing requirements for the past 90 days
. Yes
☒
No
☐Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation
S-T
(§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes
☒
No
☐Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a
non-accelerated
filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule12b-2
of the Exchange Act.| Large accelerated filer | ☐ | Accelerated filer | ☐ | |||
| Non-accelerated filer | ☒ | Smaller reporting company | ☒ | |||
| Emerging growth company | ☒ | |||||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Securities Act
. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report
. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule
12b-2
of the Act). Yes ☐ No
☒
The aggregate market value of the voting common shares held
by non-affiliates of
the registrant was approximately$193,257,000, computed by reference t
o the closing sale price of the Common Stock on The Nasdaq Capital Market on June 30, 2021, the last trading day of the registrant’s most recently completed second fiscal quarter.The number of shares of the registrant’s Common Stock outstanding as of March 15, 2022 was
122,839,613.
DOCUMENTS INCORPORATED BY REFERENCE
None.
| Auditor Firm Id: 100 | Name of Auditor: WithumSmith+Brown, PC | Auditor Location: New York, New York |
EXPLANATORY NOTE
The registrant, a Delaware corporation formerly known as Environmental Impact Acquisition Corp. (“”), was incorporated on July 2, 2020 as a special purpose acquisition company, a type of blank check company formed for the purpose of effecting a merger, capital stock exchange, asset acquisition, stock purchase, reorganization, or other similar business combination with one or more target businesses. On February 2, 2022, a date subsequent to the end of the fiscal year for which this Annual Report on Form”) is being filed, ENVI consummated the Business Combination (as defined below). In accordance with the rules of the Securities and Exchange Commission (the “”), because the Business Combination was consummated after December 31, 2021, the end of ENVI’s fiscal year, this Annual Report provides the consolidated financial statements of ENVI for the year ended December 31, 2021 and the period from July 2, 2020 (inception) to December 31, 2020, which are discussed in “.”
ENVI
10-K
(the “Annual Report
SEC
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
On January 19, 2021, ENVI consummated its initial public offering (“”) of 20,700,000 units (the “”), including 2,700,000 Units issued to the underwriters upon full exercise of their over-allotment option. Each Unit consisted of one share of ENVI’s Class A common stock, par value $0.0001 per share (“”), and”), with each whole warrant entitling the holder thereof to purchase one share of Class A Common Stock for $11.50 per share. The Units were sold at a price of $10.00 per Unit, generating gross proceeds to ENVI of $207.0 million. Simultaneously with the closing of the IPO, ENVI completed the private sale of an aggregate of 2,000,000 warrants (the “”) at a purchase price of $1.00 per Private Placement Warrant, generating gross proceeds of $2.0 million. At the closing of the IPO, ENVI also issued 600,000 Private Placement Warrants to its sponsor and 50,000 Private Placement Warrants to each of its three independent directors. A total of $207.0 million, comprised of $206,750,000 of the proceeds from the IPO and $250,000 of the proceeds of the sale of the Private Placement Warrants, was placed in a U.S.-based trust account (the “”) maintained by Continental Stock Transfer & Trust Company, acting as trustee.
IPO
Units
ENVI Class
A Common Stock
one-half
of one redeemable warrant (“warrant
Private Placement Warrants
trust account
On August 9, 2021, ENVI entered into a Business Combination Agreement (the “”), by and among ENVI, Honey Bee Merger Sub, Inc., a Delaware corporation and wholly owned subsidiary of ENVI (“”), and GreenLight Biosciences, Inc., a Delaware corporation (“”). On February 2, 2022 (the “”), pursuant to the terms of the Business Combination Agreement, Merger Sub merged with and into GreenLight, with GreenLight surviving the merger as a wholly owned subsidiary of ENVI (the “” or “”). In connection with the consummation of the Merger on the Closing Date, ENVI changed its name to GreenLight Biosciences Holdings, PBC (“”) and became a public benefit corporation.
Business Combination Agreement
Merger Sub
GreenLight
Closing Date
Merger
Business Combination
New GreenLight
In accordance with the terms and subject to the conditions of the Business Combination Agreement, at the effective time of the Merger (the “”), each outstanding share of capital stock of GreenLight (other than treasury shares) was exchanged for shares of common stock, par value $0.0001 per share, of New GreenLight (“”), and outstanding GreenLight options and warrants to purchase shares of capital stock of GreenLight (whether vested or unvested) were converted into comparable options (the “”) and warrants to purchase shares of New GreenLight Common Stock, in each case, based on an implied GreenLight fully diluted equity value of $1.2 billion. In connection with the consummation of the Business Combination, all of the issued and outstanding shares of ENVI Class A Common Stock and all of the issued and outstanding shares of ENVI Class B common stock, par value $0.0001 per share (“”), became shares of New GreenLight Common Stock.
Effective Time
New GreenLight Common Stock
Rollover Options
ENVI Class
B Common Stock
In connection with the Business Combination, New GreenLight completed the sale and issuance of 12,425,000 shares of New GreenLight Common Stock in a private placement at a purchase price of $10.00 per share pursuant to subscription agreements (the “”) that had been entered into between
Subscription Agreements
i
New GreenLight and certain institutional accredited investors (the “”) either concurrently with the execution of the Business Combination Agreement or subsequently in November 2021 (the “”).
PIPE Investors
PIPE Financing
Also in connection with the Business Combination, 19,489,626 shares of ENVI Class A Common Stock were redeemed for an aggregate payment of approximately $194.9 million. The redemption price was paid from the trust account, the remaining balance of the trust account was disbursed to New GreenLight, and the trust account was closed.
As a result of the Business Combination, GreenLight is considered the accounting predecessor of New GreenLight. The audited consolidated financial statements of GreenLight as of and for the years ended December 31, 2021 and 2020 have been filed as Exhibit 99.1 to the Current Report on Form
8-K
filed by New GreenLight on the date hereof. For filings made after the Closing Date (other than this Annual Report), the consolidated financial statements of GreenLight will be the consolidated financial statements of New GreenLight.Unless otherwise stated or the context indicates otherwise, the financial information contained in this Annual Report, other than the consolidated financial statements and “”, are those of GreenLight.
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
ii
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This annual report includes forward-looking statements regarding, among other things, the business and financial plans, strategies and prospects of New GreenLight. These statements are based on the beliefs and assumptions of the management of New GreenLight. Although New GreenLight believes that the plans, intentions and expectations reflected in or suggested by these forward-looking statements are reasonable, it cannot assure you that it will achieve or realize these plans, intentions or expectations. Generally, statements that are not historical facts, including statements concerning possible or assumed future actions, business strategies, events or results of operations, and any statements that refer to projections, forecasts, or other characterizations of future events or circumstances, including any underlying assumptions, are forward-looking statements. These statements may be preceded by, followed by or include the words “believes”, “estimates”, “expects”, “projects”, “forecasts”, “may”, “might”, “will”, “should”, “seeks”, “plans”, “scheduled”, “possible”, “anticipates”, “intends”, “aims”, “works”, “focuses”, “aspires”, “strives” or “sets out” or similar expressions. Forward-looking statements are not guarantees of performance. Forward-looking statements involve a number of risks, uncertainties (many of which are beyond the control of New GreenLight) or other factors that may cause actual results or performance to be materially different from those expressed or implied by these forward-looking statements. You should not place undue reliance on these statements, which speak only as of the date these statements were made. These risks and uncertainties include, but are not limited to, the following risks, uncertainties (some of which are beyond our control) or other factors:
| • | the anticipated need for additional capital to achieve New GreenLight’s business goals; |
| • | the need to obtain regulatory approval for New GreenLight’s product candidates; |
| • | the risk that preclinical studies and any ensuing clinical trials will not demonstrate that New GreenLight’s product candidates are safe and effective; |
| • | the risk that New GreenLight’s product candidates will have adverse side effects or other unintended consequences, which could impair their marketability; |
| • | the risk that New GreenLight’s product candidates do not satisfy other legal and regulatory requirements for marketability in one or more jurisdictions; |
| • | the risks of enhanced regulatory scrutiny of solutions utilizing messenger ribonucleic acid (“ mRNA ”) as a basis; |
| • | the potential inability to achieve New GreenLight’s goals regarding scalability, affordability and speed of commercialization of its product candidates; |
| • | the potential failure to realize anticipated benefits of the Business Combination or to realize estimated pro forma results and underlying assumptions; |
| • | changes in the industries in which New GreenLight operates; |
| • | changes in laws and regulations affecting the business of New GreenLight; |
| • | the potential inability to implement or achieve business plans, forecasts, and other expectations; |
| • | the potential inability to maintain the listing of New GreenLight’s securities with Nasdaq; |
| • | the outcome of any legal proceedings that may be instituted against New GreenLight related to the Business Combination; |
| • | unanticipated costs related to the Business Combination, which may reduce available cash; |
| • | the effect of the Business Combination on New GreenLight’s business relationships, operating results, and business generally; |
| • | risks that the Business Combination disrupts current plans and operations of New GreenLight; and |
iii
| • | other factors detailed in this annual report under the section entitled “ Risk Factors |
The risks described under the heading “” in this annual report are not exhaustive. New risk factors emerge from time to time, and it is not possible to predict all such risk factors, nor can we assess the impact of all such risk factors on the business of New GreenLight or the extent to which any factor or combination of factors may cause actual results to differ materially from those contained in any forward-looking statements. All forward-looking statements attributable to New GreenLight or to persons acting on our behalf are expressly qualified in their entirety by the foregoing cautionary statements. Some of these risks and uncertainties may in the future be amplified by
Risk Factors
the COVID-19 pandemic,
and there may be additional risks that New GreenLight considers immaterial or which are unknown. New GreenLight does not undertake any obligation to update or revise any forward-looking statements, whether as a result of new information, future events or otherwise, except as may be required under applicable securities laws.TRADEMARKS
This document contains references to trademarks, trade names and service marks belonging to other entities. Solely for convenience, trademarks, trade names and service marks referred to in this annual report on Form 10-K may appear without the
®
or TM symbols, but such references are not intended to indicate, in any way, that the applicable owner will not assert, to the fullest extent under applicable law, its rights to these trademarks and trade names. We do not intend our use or display of other companies’ trade names, trademarks or service marks to imply a relationship with, or endorsement or sponsorship of us by, any other companies.iv
SELECTED DEFINITIONS
Unless otherwise stated in this Annual Report or the context otherwise requires, references to:
| • | “ Alternative Forum Consent |
| • | “ Business Combination |
| • | “ Business Combination Agreement |
| • | “ Business Combination Marketing Agreement |
| • | “ Bylaws |
| • | “ Canaccord |
| • | “ Charter |
| • | “ Closing |
| • | “ Closing Date |
| • | “ Continental |
| • | “ DGCL |
| • | “ Effective Time |
| • | “ ENVI |
| • | “ ENVI Board |
| • | “ ENVI Class A Common Stock |
| • | “ ENVI Class B Common Stock |
| • | “ ENVI common stock |
| • | “ ENVI Units one-half of one redeemable warrant entitling the holder of such warrant to purchase one share of ENVI Class A Common Stock at a price of $11.50 per share; |
| • | “ Exchange Act |
| • | “ Former Bylaws |
v
| • | “ Former Charter |
| • | “ Former Organizational Documents |
| • | “ GreenLight |
| • | “ GreenLight 2012 Equity Plan |
| • | “ GreenLight Common Stock |
| • | “ GreenLight Financial Statements 8-K filed by New GreenLight on the date hereof and which are incorporated herein by reference; |
| • | “ GreenLight MD&A Management’s Discussion and Analysis of Financial Condition and Results of Operations of GreenLight 8-K filed on the date hereof and which is incorporated herein by reference; |
| • | “ GreenLight Preferred Stock |
| • | “ GreenLight Series A Preferred Stock A-1 Preferred Stock, SeriesA-2 Preferred Stock and SeriesA-3 Preferred Stock, in each case with a par value $0.001 per share, of GreenLight; |
| • | “ GreenLight Series B Preferred Stock |
| • | “ GreenLight Series C Preferred Stock |
| • | “ GreenLight Series D Preferred Stock |
| • | “ GreenLight Shares |
| • | “ GreenLight stockholders |
| • | “ HB Strategies |
| • | “ initial public offering IPO |
| • | “ Initial Stockholders |
| • | “ Insider Warrants |
vi
| • | “ Instruments |
| • | “ Investment Agreement |
| • | “ Merger |
| • | “ Merger Sub |
| • | “ Nasdaq |
| • | “ New GreenLight |
| • | “ New GreenLight Board |
| • | “ New GreenLight Common Stock |
| • | “ New GreenLight Equity Plan |
| • | “ New GreenLight ESPP |
| • | “ PBC |
| • | “ PBC Purpose |
| • | “ PIPE Financing |
| • | “ PIPE Investors |
| • | “ PIPE Prepayment |
| • | “ Prepaying PIPE Investors |
vii
| • | “ Private Placement Warrants |
| • | “ pro forma |
| • | “ Promissory Note |
| • | “ public common stock |
| • | “ public stockholders |
| • | “ Public Warrants |
| • | “ redemption |
| • | “ SEC |
| • | “ Securities Act |
| • | “ SIIPL |
| • | “ Sponsor |
| • | “ Sponsor Warrants |
| • | “ Subscription Agreements |
| • | “ transfer agent |
| • | “ trust account |
| • | “ Warrant Subscription Agreement |
viii
PART I
ITEM 1. | Business |
GreenLight has a clear mission: To create products addressing some of humanity’s greatest challenges through the rigorous application of science.
We aim to achieve this goal through our cell-free biomanufacturing platform. This platform enables us to make complex biological molecules—nucleic acids, peptides, carbohydrates, and many others—in a manner that we believe will allow us to manufacture high-quality products at a lower cost than traditional methods using fermentation. We are using this platform to develop and commercialize products that, if they receive appropriate regulatory approvals, address agricultural, human health and animal health issues.
Humanity faces numerous challenges. There are more than seven and a half billion people sharing the diminishing resources of Earth. This growing population needs to produce more food with the same amount of land and, at the same time, honor the global desire—and increasing technical need—to replace chemical pesticides. Not only are these pesticides facing increased consumer opposition and threat of outright bans due to environmental damage, many are losing their effectiveness.
More than half the world’s population now lives in cities, breathing the same air that carries pathogens and causes infections. Humanity needs to adapt and tackle pandemics both for those who have and for those who do not have access to good health care around the planet.
To address these issues, we need to develop high-quality, cost-effective products that can be deployed widely, including to developing countries. We believe RNA can be the critical aspect to these products.
Ribonucleic acid, or RNA, recently gained broad global prominence as
the COVID-19 pandemic
swept through the world’s population, prompting messenger RNA, or mRNA, vaccines to move from a scientific theory to a medical reality. Vaccines made using mRNA proved among the fastest to develop and the easiest to update for newer strainsof COVID-19.
While the fast rollout of mRNA vaccines helped change the course of the pandemic, this is just one part of the story. The full potential for RNA in human health has not yet been been realized. Beyond human
health, RNA-based technology
can also be deployed to address other global issues, including agricultural needs for crop protection.Our technology platform, which was initially developed to produce agricultural crop protection products and is protected by patents and
know-how,
is capable of synthesizing building blocks (nucleotides), building tools (enzymes), and instructions (DNA templates) to make dsRNA within an integrated process. The manufacturing processknow-how
that we gained from our experience making dsRNA allows us to understand some of the key aspects of producing mRNAs. For more information on our manufacturing platform and technology, see“Item 1. Business – Our Manufacturing Platform.”
We have several dsRNA-based products in our agricultural pipeline that, if commercialized, we believe can change the way in which farmers protect crops, allowing them to better utilize the land dedicated to agriculture and produce foods with less or no pesticide residue. One of these products, Calantha which is designed to manage Colorado potato beetles, has been submitted to the EPA for approval. Our other dsRNA-based agricultural products are in various earlier stages of development as compared to Calantha, our Colorado potato beetle product, ranging from proof of concept in the lab to proof of technology in the greenhouse and proof of scale in the field. See “” for additional information on the development process. In order to commercialize a product for the U.S. agricultural market, we must complete specified toxicology studies, submit a registration dossier to the EPA
Item 1. Business — Plant Health Product Pipeline — Process for developing new products
1
demonstrating that the product does not pose unreasonable risks to human health or the environment, respond adequately to any deficiencies identified by the EPA through its risk assessment process and obtain the EPA’s approval of our labeling. The EPA must also establish a tolerance level for the product or issue a tolerance exemption. We must separately obtain any applicable state or foreign regulatory approvals. For more information regarding the regulatory process, “” and “”.
Item 1.
Business –
Government Regulation – Agricultural Products
Item 1A.
Risk Factors – Risks Related to Our Plant Health Program
We are also in
pre-clinical
development ofRNA-based
vaccines directed at arresting the damage of the current viral pandemic and addressing emerging pathogens. The first candidate in this product pipeline that we hope to bring to market is aCOVID-19
vaccine, which is currently being tested on animals in toxicity studies in anticipation of filing a Clinical Trials Application, or CTA, with the South African Health Products Regulatory Authority, or SAHPRA, which, if approved, will allow clinical testing on human subjects. Depending on the results of any clinical testing in South Africa, we may decide to file an Investigational New Drug, or IND, application with the FDA for additional testing of ourCOVID-19
vaccine candidate. Other product candidates in the human health pipeline have yet to reach thePre-IND
phase. To get to thePre-IND
phase for our other product candidates in our human health pipeline, we must successfully design and test the product candidates in animal models, achieve positive results, select the product candidates to progress toIND-enabling
toxicology studies, develop chemistry, manufacturing, and controls protocols and create a development plan to discuss with the FDA as part ofpre-IND
consultations.AN INTRODUCTION TO RNA
RNA is present in all known life forms and plays an essential role in numerous biological processes but primarily provides a template by which proteins are constructed. In some organisms RNA can be the mechanism by which those templates are stored, but, in higher forms of life, it translates the code stored in the form of DNA and provides the template to convert that code into proteins by transcribing the DNA. Consequently, RNA molecules are being studied as potential products in many fields, such as agriculture (for pest control), animal health, and human health (messenger
RNA-based
vaccines and gene therapies).RNA can be transformative for human health and plant health:
| • | Human and animal health—where messenger RNA, or mRNA, can be used to express proteins which form the basis of vaccines as well as other therapies. |
| • | Plant health—where dsRNA can be leveraged to regulate the expression of a target protein by interfering with its message. Such RNA-mediated interference can form the basis for highly targeted pesticides or protection against parasites. |
RNA in agriculture
New crop-protection strategies are urgently needed as pests become resistant to existing pesticide products. Many existing products are also being limited through primary regulatory action (government regulations) or secondary regulations (food chain regulation) because of concerns about their effects on humans or the environment, with environmental concerns
including off-target toxicity
and long-term effects on crops, soil, and water. Together, these factors spur the need to develop alternative crop-protection products with new modes of action and improved safety profiles.Double-stranded RNA products in agriculture exploit a natural biological process called RNA interference (RNAi). This a biological process found in many eukaryotic organisms, which break down dsRNA that has been taken into a cell into short fragments known as
either micro-RNA or
small interfering RNA (siRNA). The presence of these small RNA fragments can lead to the degradation of the corresponding mRNA, thereby limiting or stopping the synthesis of protein specific to a particular harmful pest insect.2
RNA-based
pesticides may be able to give us more environmentally friendly ways to protect crops and beneficial insects while effectively stopping harmful pests. Much of our ability to design products that are intended to improve the environmental profile associated with crop protection products relies on our ability to design an RNA sequence that is only found in the organism(s) that we desire to manage. In this design process, we will compare the genome of our target species to the genome of other species that mayco-exist
with it, including humans. The goal is to have no overlap with the genome of other organisms by which our products would harm those organisms.RNA in human health
Messenger RNA’s features make it broadly valuable for human health. It is well known that mRNA has been used to make some of the most
effective COVID-19 vaccines
and that these vaccines have been developed quickly, which is critical for a pandemic response. More than one billion doses ofmRNA COVID-19 vaccine
have been produced. However,beyond COVID-19 vaccines,
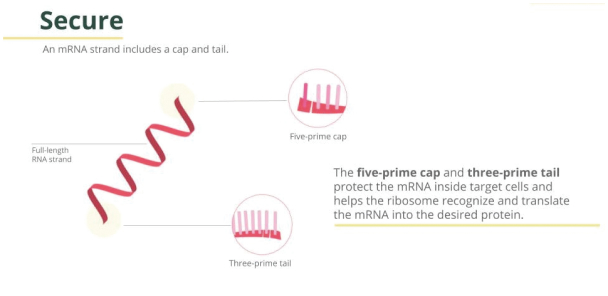
mRNA’s features make it valuable for other vaccines and therapies. We are working on using mRNA in multiple vaccines and therapeutic applications such as an influenza vaccine, a sickle cell gene therapy and a shingles vaccine.DNA encodes the instructions for life to function. DNA is transcribed into mRNA in the cellular nucleus, and subsequently this mRNA is translated into proteins in the cellular cytoplasm. Instructions to make proteins that help perform many critical functions are transcribed from the DNA to mRNA. The mRNA consists of four ribonucleosides (Adenine, Guanine, Cytosine, and Uracil), the sequence of which determines the structure of the proteins encoded. Synthetic mRNA can be produced in manufacturing facilities for delivery into the cellular cytoplasm, enabling the cells to produce proteins as vaccines or for therapy.
mRNA has many advantages:
| • | Wide range of applications: mRNA can produce any encoded protein (intracellular, membrane-bound, or secreted), giving it many uses in vaccines, gene therapy, or for therapeutic proteins. |
| • | Transient expression: The body has mechanisms to degrade mRNA, allowing for repeat dosing and a dose response which can be tailored for the needs of the pharmaceutical product. |
| • | Fast development: Relatively simple changes to the mRNA molecule are needed to produce different therapeutic proteins, enabling a fast turnaround from gene selection to product with little need for manufacturing changes. For instance, if a booster vaccine is needed for a new variant, no changes will need to be made except to the mRNA sequence itself. |
| • | Flexible manufacturing: A single manufacturing facility can produce different vaccines and therapies, as the process is essentially the same regardless of the product. |
Until recently, it was very difficult to stabilize mRNA, understand its interaction with the human immune system, or deliver it in vivo. Addressing these challenges has allowed the development of the RNA industry and its rapid deployment as a global healthcare product.
THE IMPACT OF OUR
HIGH-QUALITY, LOW-COST RNA
We currently make multiple forms of dsRNA at a rate of 2,000 liters per batch using our cell-free manufacturing platform. We have increased our production rate from microliters to milliliters to liters to our current
2,000-liter
capacity with no material impact on quality or process yields. We believe our expertise and proprietary technology will allow us to increase batch sizes to 10,000 liters and beyond, which will allow us to reduce theper-liter
cost of our dsRNA products.3
Alternative RNA production methods are generally slow to develop and more expensive:
| • | Cell-based fermentation does not achieve the quality required for human health uses or the cost considerations for broadacre coverage in agriculture applications. |
| • | Conventional cell-free processes, such as in vitro transcription (IVT), are cost prohibitive for agricultural applications and require complex specialty input supply chains. |
GreenLight’s manufacturing platform uses:
| • | a proprietary cell-free methodology that enables production at less than $1/gram for the production of technical grade active ingredient dsRNA. |
| • | a flexible architecture that accommodates the manufacturing of a wide variety of products. |
For more information on our manufacturing platform and technology see the section titled “.”
Item 1. Business—Our Manufacturing Platform
OUR BUSINESS MODEL AND GROWTH STRATEGY
Given the advantages of our platform, we aim to make the benefits of RNA, and other biologics, accessible to everyone.
In human health, we are developing vaccines and RNA therapeutics to alleviate or cure critical diseases facing patients worldwide. In agriculture, we are developing products that promote sustainability and supplement or replace traditional pesticides and fungicides with RNA in farmers’ crop-protection programs.
Our platform gives rise to three distinct capabilities that underpin a sustainable business model and bring capabilities normally used in advanced pharma discovery to agriculture and human health:
| • | Identification: Machine learning and proprietary algorithms are key tools as we work to identify the best gene target candidates. We become more efficient and innovative as we accumulate data, and our algorithms learn. |
| • | Develop and optimize: We run parallel trials on thousands of distinct RNA sequences to design our agricultural products, which gives us many more opportunities to develop the best products. |
| • | Manufacturing: We can produce dsRNA products through our proprietary cell-free system. Our current production capacity is 2,000 liters per batch, and we are planning to build the capacity to produce dsRNA at a rate of at least 10,000 liters per batch. Production at larger capacities will allow us to achieve economies of scale by reducing labor costs and the fixed costs that we allocate to each liter of RNA that we produce. |
In the next five years, our pipeline includes seven agricultural products planned for launch and five human health products with clinical milestones, including Phase I clinical trials.
We anticipate this pipeline will demonstrate:
| • | Fast development of agricultural products.Calantha will, if approved in 2022, have taken four years from start to market compared to a typical 10-year cycle at major agribusinesses. |
| • | Rapid integration of acquisitions. We acquired Bayer’s topical RNA treatment for honeybees in December 2020. By May 2021, we were conducting further field trials and intend to be ready for regulatory submission in 2022. |
| • | Validation of our mRNA platform. We are working toward clinical proof of concept of our COVID-19 and influenza mRNA vaccines. |
4
| • | Innovative approaches to gene editing. We have the potential to tackle grave diseases such as sickle cell, for which we received a $3.3 million grant from the Bill & Melinda Gates Foundation. |
| • | Expansion of production capabilities. Our Rochester RNA manufacturing facility can produce 500 kg of dsRNA per year with the capability to expand to 1,000 kg. It currently provides samples for our field trials. |
These factors allow us to access the following major markets (with estimated total addressable market size in parentheses):
| • | Insecticides ($17 billion) |
| • | Fungicides ($16.5 billion) |
| • | Vaccines ($93 billion) |
| • | Gene therapies ($3 billion) |
Our growth strategy in plant health is to pursue significant market opportunities where RNA has the greatest potential to provide growers with improved pest control and the sustainable nature of our products (e.g., benefits to honeybees and low to no residue) delivers the most impact for society and aligns most closely with macro trends from consumers and regulators. When we use the term ‘sustainable,’ we refer to our efforts to align economic development with environmental protection and human well-being as well as our anticipated obligations as a Public Benefit Corporation under § 362(a) of the Delaware General Corporation Law.
For our plant health products, we define total addressable market as the global revenue opportunity available to pesticide solutions controlling a target pest or disease. In most instances, we do this by defining a relevant active ingredient market for the crop or crops where we intend to market our products and then making an assumption as to the percentage of that market that is spent on controlling the target pest or disease. We use data from AgBioinvestor and FAOSTAT (a database run by the Food and Agriculture Organization of the United Nations), and data purchased from third party consultants is used to quantify the market and underpin assumptions. In order to address the total insecticide and fungicide markets we identify pests or diseases in that market, develop targeted dsRNA sequences for them and attempt to develop the best delivery mechanism for that dsRNA. Over time, we intend to expand beyond RNA, building on our capabilities. In human health, we intend to pursue markets where RNA can provide better products (faster, cost effective or more efficacious) to improve standards of care for patients.
Planned products and milestones
Agricultural programs we currently have planned for launch in the next five years include protection against:
| • | Colorado potato beetle, 2022 |
| • | Varroa mite, 2024 |
| • | Botrytis, 2025 |
| • | Fusarium, 2025 |
| • | Powdery mildew, 2025 |
| • | Diamondback moth, 2026 |
| • | Two-spotted spider mite, 2026 |
Key planned human health clinical milestones in the next five years include Phase I clinical trials currently targeted for:
| • | COVID-19 vaccine, 2022 (currently in animal toxicity studies) |
5
| • | Seasonal flu vaccine, late 2022/early 2023 (currently in pre-toxicity study development) |
| • | Shingles, 2024 (currently in early stages of concept evaluation) |
| • | Supra-seasonal flu, 2024 (currently in early stages of concept evaluation) |
| • | Antibody therapy, 2024 (currently in early stages of concept evaluation) |
| • | Sickle cell disease product concept, 2025 (currently in early stages of concept evaluation) |
Our
Covid-19
vaccine has successfully completed preclinical testing and we are preparing a Clinical Trials Application, or CTA, with the South African Health Products Regulatory Authority, or SAHPRA, which, if approved, will allow clinical testing on human subjects. Depending on the results of any clinical testing in South Africa, we may decide to file an Investigational New Drug, or IND, application with the FDA for ourCOVID-19
vaccine candidate. Our other product candidates in the human health pipeline have yet to reach thePre-IND
phase. To get to thePre-IND
phase for our other product candidates in our human health pipeline, we must design and test the product candidates in animal models, select the product candidates to progress toIND-enabling
toxicology studies, develop chemistry, manufacturing, and controls protocols, and create a development plan to discuss with the FDA as part ofpre-IND
consultations.In order to begin any Phase I clinical trials, we must first successfully complete the toxicity study for the product candidate, submit an IND application to the FDA, which will include the scope of our proposed Phase I clinical trial, and satisfy any conditions the FDA may require for the IND to become effective. Additionally, in order to begin our Phase I clinical trials, we must first produce the Phase I clinical drug substance for each of the product candidates, which will require us to complete the.” Our Burlington facility completed the production of the drug substance for the Phase I clinical trial of our COVID-19 vaccine product candidate. Fill and finish manufacturing is planned for the first and second quarter of 2022.
start-up
of the two clean room suites that we recently leased in Burlington, Massachusetts, including compliance with applicable GMP regulations and conformity to our chemistry, manufacturing and controls protocols. Our Burlington facility is currentlyGMP-ready.
See “Item 1A. Risk Factors — Risks Relating to Our Manufacturing Platform
OUR MANUFACTURING PLATFORM
Our platform, developed through 13 years of research and technology development, is protected by foundational patents and
know-how
that address barriers cell-free technologies have faced for many years.Biologic production through living cells faces a range of constraints. These include the cell’s priority for self-preservation, which fights against RNA production, reducing yield and quality.
Conventional cell-free production breaks open the cells and removes the need to balance bioprocessing against self-preservation. But energy management in this method limits yield and quality, making RNA production prohibitive for many agricultural applications in terms of cost, scale, and speed.
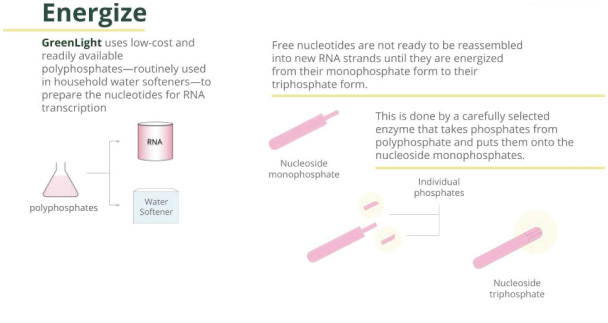
Our proprietary cell-free process regenerates the energy needed for bioprocessing using ingredients that can include polyphosphates and enzymes.
Each step in the GreenLight bioproduction processes has been developed or selected with cost and functionality in mind.
| • | The key raw material for dsRNA can be obtained in large quantities from such sources as industrial fermentation processes (e.g., derived from yeast). |
| • | Our proprietary process allows us to energize naturally occurring nucleoside monophosphates at low cost using inorganic polyphosphate, which is readily available and affordable. |
6
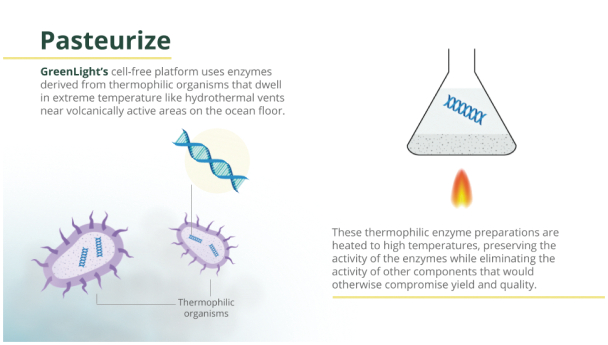
| • | Thermophilic enzymes are employed to facilitate the production of high-energy nucleotides. The utilization of thermally stable enzymes allows high temperature to be incorporated in their preparation, providing a way to mitigate undesirable contaminating activities (e.g., RNA-degrading enzymes,DNA-degrading enzymes, nucleotide-degrading/altering enzymes, protein-degrading enzymes) from entering the RNA synthesis portion of the process and affecting quality and yield. |
| • | We believe our process know-how and the technology we developed can be leveraged for our mRNA platform. |
Overview of manufacturing process for agriculture
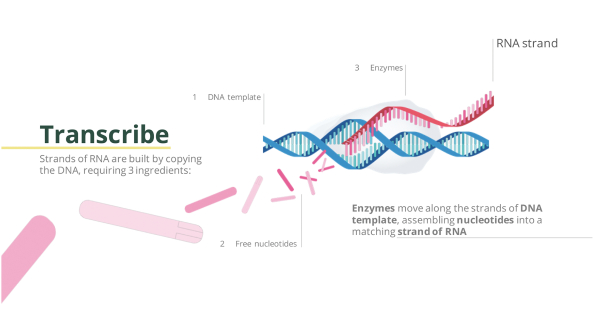
Our proprietary dsRNA manufacturing process for agriculture begins with a cellular RNA (e.g., from yeast).
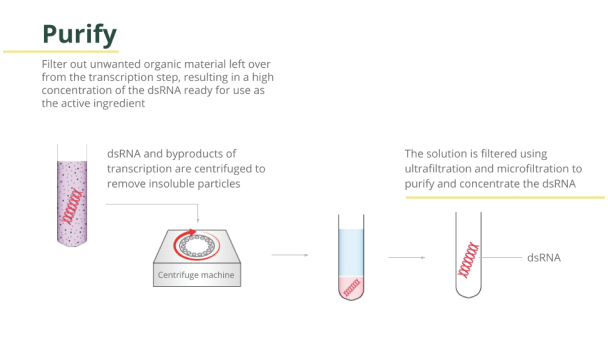
This is then broken up (depolymerized) into RNA building blocks (commonly known as NMPs) using a nuclease enzyme. Our cell-free production process uses carefully selected enzymes to energize and polymerize the building blocks into a desired RNA according to a corresponding DNA template.
Cell-free production of dsRNA for agriculture
While the advantages of dsRNA for agriculture have been known for some time, production cost has been a barrier.
The dsRNA production process, simplified

7
8
The outcome from our system is fast, with reaction times of two hours. We have successfully increased production from 50 microliters to more than 1,000 liters without significant loss in performance.
Overview of manufacturing process for human health
We have deep understanding and expertise in RNA manufacturing, design, and analysis. The many years of experience from our dsRNA platform are applicable to our human health mRNA platform. We are continually leveraging what we learn and applying it to mRNA production for human health applications.
State-of-the art
Covid-19
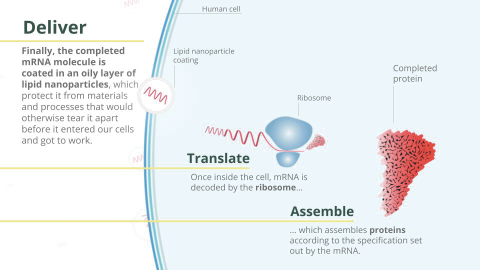
mRNA vaccines) employs in vitro transcription (IVT). The process depends on a ready supply of highly purified reagents, including chemically produced nucleoside triphosphates (NTPs), an RNA polymerase enzyme, and a DNA template. RNA is synthesized, capped, and tailed for protein translation and encapsulated in lipid nanoparticles (LNP) for delivery to target cells in the patient. Importantly, mRNA used for human health requires purification steps to reach the highest quality levels expected by regulatory agencies.Encapsulation
Our current mRNA drug product is based on lipid nanoparticles that encapsulate mRNA molecules, protecting them from degradation. Those nanoparticles enable mRNA uptake into the cells so that the mRNA can be used to express the protein of interest.
Our current nanoparticles are made of four lipids: the ionizable lipid that drives encapsulation and release of mRNA, two “helper” lipids that mainly provide stability to the particle itself, and a polyethylene-glycol lipid that prevents particle aggregation as well as opsonization once those particles are injected in the bloodstream.
The manufacturing process
for mRNA-LNP involves
two liquid streams colliding at high velocity ina jet-mixing chamber.
One of the streams contains the lipids in organic solvents and the other stream contains the mRNA in acidified water. The mixing at high velocity reduces solubility of the lipids so that homogeneous nanoparticles are formed around a core made of mRNA and ionizable lipid.9
After the mixture is quenched to stop particle growth, the organic solvent is removed, the pH is neutralized, and
the mRNA-LNP is
concentrated.A cryoprotectant is added at the end of the process before the product is sterile-filtered and stored in ultra-cold conditions.
10
Supply for research and development
We have a team dedicated to the manufacturing of materials for discovery and preclinical research in Massachusetts. Our team produces 1 to 20 mg of mRNA with a turnaround time of a few weeks, and we have
technology-transferred in-house LNP
manufacturing capability to support preclinical and clinical studies.Supply for clinical trials
In August 2021, we began activating a manufacturing facility in Massachusetts that is capable of producing material for clinical trials and is implementing current good manufacturing practice (cGMP) systems to support the use of these materials in human trials. The first clinical materials to be produced in this facility, which were for our
COVID-19
vaccine candidate, were manufactured in 2021, and we expect to follow with production for other programs entering the clinical phase. We have also produced mRNA and LNP formulations forIND-enabling
toxicology studies under Good Laboratory Practice (GLP) procedures. We contract specialized third parties that meet our requirements and are experienced in cGMP fill-finish operations.Our manufacturing for agriculture: Rochester (dsRNA)
Our dsRNA manufacturing facility in Rochester, New York, is designed for process development while generating samples for research and market development with a 1,000 kg dsRNA production design basis. The facility has a raw material storage and handling area, high
bay wet-processing area
with floor drains, two,1,200-liter
fermenters,2,000-liter
cell-free reactors, NMP preparation tanks, formulation, packaging, development laboratory, analytical laboratory, loading dock, and cold storage areas. This plant currently has 15 process engineers, technicians, research associates, and quality-control personnel for commercial production of plant health products.In the third quarter of 2021, we increased production of dsRNA from microliter, milliliter and liter quantities in the lab to 2,000 liters per batch using a single bioreactor installed at our Rochester facility with no material impact on quality or process yields. We estimate that we can currently produce 500 kg of dsRNA per year with this capacity. We have also installed a second
2,000-liter
bioreactor at our Rochester facility as part of our plan to increase manufacturing capacity. If brought online, this bioreactor would double our capacity to 1,000 kg of dsRNA per year. If we obtain the appropriate regulatory approvals to commercialize our products as we project in our agricultural product pipeline, we expect that this capacity will enable us to meet our agricultural product needs throughmid-2024.
We are currently designing an expansion of our Rochester facility to further increase our manufacturing capacity of dsRNA for agricultural use and have recently leased an additional 5,577 square feet of laboratory space in Rochester for this added capacity.11
Our manufacturing for human health (mRNA)
We currently make mRNA at our cleanroom facility in Burlington, Massachusetts. We have scaled our production to approximately three liters and plan to use a contract development and manufacturing organization, or CDMO, to produce mRNA in larger quantities.
We have agreements to produce early clinical materials for the
COVID-19
program, which are expected to be filled, labeled, and packaged for the clinic by contract manufacturing organizations.Our Burlington facility completed the production of the drug substance for the Phase I clinical trial of our COVID-19 vaccine product candidate. Fill and finish manufacturing is planned for the first and second quarter of 2022.
We are implementing cGMP systems to support clinical production using our process.
We produced our first
3-liter
clinical batch using our proprietary process and will use the wild-type WuhanCOVID-19
strain. This material will follow the same supply chain but will use an IVT production process currently in development, akin to existing vaccines, in the upstream.The downstream purification process consists of filtration and chromatography steps. The purified material is formulated in a lipid nanoparticle system. The material is then sent to a contract manufacturer to fill into vials and is stored frozen.
In November 2021, we engaged Samsung Biologics Co., Ltd. (“basis for each year under that agreement, for our minimum purchase commitments, as determined pursuant to the terms of the Samsung Agreements. Based on our minimum purchase commitments, we expect to pay Samsung a minimum of approximately $11.5 million in service fees under the Samsung Agreements, excluding the cost of raw materials, which we must supply to Samsung separately. These fees include initial technology and analytical method transfer fees, process development and
Samsung
”) as a contract development and manufacturing organization for our mRNACOVID-19
vaccine pursuant to a Master Services Agreement (the “MSA
”) and a Product Specific Agreement (the “PSA
”, and together with the MSA, the “Samsung Agreements
”). Under the Samsung Agreements, Samsung will perform pharmaceutical development and manufacturing services for us over a period of years at its South Korean facility in exchange for service fees. Under these agreements, we must purchase certain minimum quantities of drug products. We agreed that, if we enter into a purchase agreement for commercial quantities of drug product, we will pay Samsung, on a minimumtake-or-pay
scale-up
fees, process characterization fees, an annual project management fee, andper-batch
engineering and cGMP run fees. Based on our current schedule, we expect to incur the substantial majority of these expenses in 2022 and a portion in 2023. If we move to commercial production, the agreement provides for additional process validation, inspection, cleaning, stability testing and commercial production fees, most of which would be incurred on aper-batch
basis.The Samsung Agreements will terminate on December 31, 2026, unless earlier terminated or extended in accordance with their terms. If we terminate the Samsung Agreements, we will generally be responsible for paying the purchase price for our aggregate product commitment for the remainder of the term, less any amounts we have already paid. Samsung agreed that, at or before the end of the term of the Samsung Agreements, it will assist us to transfer the commercial scale manufacturing process to a facility designated by us. The Samsung Agreements impose limits on Samsung’s liability to us for breaches of the agreements.
HUMAN HEALTH PRODUCT PIPELINE
Our mRNA platform consists of:
| • | The manufacturing process used to produce the product (described above) |
| • | The mRNA molecule |
| • | The delivery vehicle it uses to reach the target tissue |
12
All elements of the platform affect product characteristics, such as purity, potency, and immunogenicity, so our teams work on optimizing mRNA molecules and delivery vehicles for a given indication, and the performance and cost of the manufacturing process.
mRNA molecule design
Changes in the mRNA molecule will result in changes in the protein we are looking to express, the immune response to the product, and mRNA stability and potency.
First, we must choose the right target protein, after which an mRNA has to be designed for it. A well-designed mRNA molecule can carry instructions for the relevant protein, to be expressed efficiently and for the desired duration. The mRNA composition can be optimized to avoid undesirable immune responses while increasing protein expression, referred to as product potency.
Delivery vehicles
To facilitate the mRNA to reach its destination without degradation, we must formulate it into a delivery vehicle. The delivery system’s design can influence potency, immunogenicity, and the product’s shelf life.
One such delivery system consists of encapsulating mRNA in lipid nanoparticles (LNPs). We work with several established companies that have extensive experience in clinical LNPs for our vaccine candidates. We are able to routinely produce our mRNA-containing LNPs to support our research and development efforts. In addition, we work on stabilizing the LNPs to improve the storage conditions and shelf life of our products.
Our human health pipeline
We are currently working on two modalities:
| • | Prophylactic vaccines for infectious diseases |
| • | Gene therapies |
We are exploring ways to expand our pipeline to include additional therapeutic areas, including using antibodies, in the future.
Prophylactic vaccines for infectious diseases
The objective of a prophylactic vaccine is to expose the body to a protein which is present in the disease-causing virus or bacterium, called the antigen, so that it can generate an immune response in the absence of the pathogen and be prepared to fight the actual infection, should it occur in the future. mRNA can potentially be used to encode the antigen as a way to expose the body to a component of the pathogen, avoiding the use of a whole infectious agent.
Vaccines that use mRNAs present significant advantages compared
to non-mRNA vaccines,
including:| • | The antigen expressed is a true match to the protein present in the pathogen, thus increasing the potential for quality of the immune response as compared to vaccines produced through other methods, in which manufacturing processes may result in changes to the antigen. |
| • | The short development time from antigen selection to clinical trials makes mRNA ideal for emerging epidemics or pandemic response. This is why mRNA vaccines have been among the fastest developed for COVID-19. |
| • | The same manufacturing plant can be used to produce different mRNA vaccines. |
13
Opportunity
Immunization with prophylactic vaccines has become one of the most successful of all healthcare interventions. It is estimated that vaccines prevent 6 million deaths every year. The global vaccine market in 2019 was estimated at $33 billion, with the surge of
the COVID-19 pandemic
representing a significant growth in the market to $93 billion in 2020.Our COVID-19 vaccine
candidateUnmet need
Although a large portion of the population in high-income countries has been vaccinated
against COVID-19, widespread
vaccination is not expectedin mid- and low-income countries
until mid-2022 or
early 2023. Tokeep COVID-19 at
bay, a periodic booster may be necessary as global herd immunity is unlikely to be achieved. Current research is ongoing to determine if booster shots will be necessary to cover waning immunity or new variants.Product concept
Our COVID-19 vaccine
candidate,GLB-CoV-2-043,
a SARS-CoV-2 variant
Achievements to date and future milestones
Ourvirus (isolateprovided protection fromchallenge using percent body weight (% BW) change as a criterion. We observed a statistically significant (p < 0.0001) reduction in weight loss, compared to controls on Day 6, the peak of disease, and Day 14, the end of the challenge study.
COVID-19
vaccine candidate has successfully completed preclinical testing, and we are preparing to file a Clinical Trials Application, or CTA, with the South African Health Products Regulatory Authority, or SAHPRA, seeking approval to initiate phase 1 clinical studies in South Africa. Hamsters (16/group) were immunized at day 0 and 21 at three dose levels of 5 µg, 30 µg, and 100 µg of vaccine or controls of saline or LNP. At day 40 of the study the animals were intra-nasally challenged with liveSARS-CoV-2
USA-WA1/2020).
Animals were followed for 14 days, and their weights were taken daily. This hamster challenge study revealed that all doses ofGLB-CoV-2-043
SARS-CoV-2
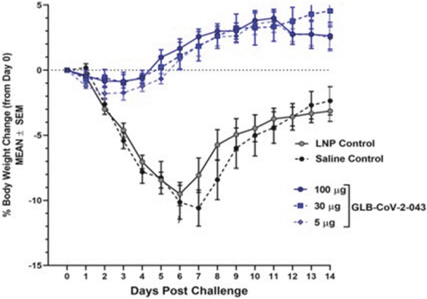
Body weight changes of vaccinated hamsters afterviral challenge,the GreenLight vaccine candidate.
SARS-CoV-2
GLB-CoV-2-043:
14
Hamsters (8/group) were immunized at day 0 and 21 at three dose levels of 5 µg, 30 µg, and 100 µg of vaccine or controls of saline or LNP (GLuc). Blood draws at day 21virus (isolate
(pre-boost:
before injection of the second vaccine dose), and day 39 (post-boost: that is, 18 days after the second dose) were tested for neutralizing antibody titers against liveSARS-CoV-2
USA-WA1/2020).
All vaccine doses tested induced significantly higher levels ofneutralization titer compared to controls, both Pre and Post-Boost. The 100 µg and 30 µg doses ofinduced statistically significantly or trending towards statistically significantly higher titers of neutralizing antibodies compared to control immunized animals. Thevaccine displayed a clear dose response after boost. These results demonstratedvaccine candidate induces high titers of functionalcapable of neutralizing virus entry into cells.
SARS-CoV-2
GLB-CoV-2-043
GLB-CoV-2-043
that GLB-CoV-2-043 mRNA
anti-SARS-CoV-2
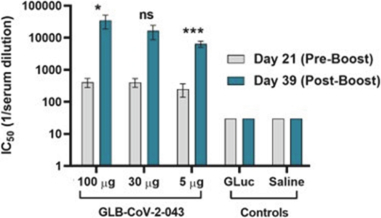
*: p<0.05
***: p<0.001
ns: p=0.0523 (not significant)
The above chart presentsserum neutralizing antibody titers (ICat day 21 (three weeks after the first vaccination dose) and at day 39 (18 days after the second vaccination dose).is the GreenLight vaccine candidate.
SARS-Cov-2
50
, or inhibition concentration at 50%) for hamsters vaccinated withGLB-Cov-2-043
GLB-COV-2043
We have had
a pre-IND consultation
with the FDA regarding our approach to IND and Phase I clinical testing. We commencedIND-enabling
toxicology studies in the third quarter of 2021. We have a license for an LNP from an established company forour COVID-19 vaccine
candidate.Upon approval of our CTA by SAHPRA, we expect to initiate a phase 1 safety and immunogenicity clinical trial in South Africa in the first half of 2022. We are also seeking partnership opportunities with a pharmaceutical company to conduct late-stage clinical trials and commercialize our vaccine. If we successfully complete our planned clinical trials, we plan to analyze the data and assess whether to submit an application package to regulatory authorities in jurisdictions outside the U.S. for emergency or full marketing authorization. It is possible that the regulatory authorities will no longer be accepting Emergency Use Authorization (EUA) submissions (or their equivalent) for
COVID-19
vaccines at that time. If that is the case, we would need to assess whether and to what extent additional data would be needed to submit a Biologics License Application (BLA), or its equivalent in other jurisdictions, for full marketing authorization.15
Our seasonal influenza vaccine candidate
Unmet need
Commercial influenza vaccines typically have a mismatch between the strains selected for the season and the strains circulating during the season, because selection occurs six months before influenza season—given the time required to manufacture vaccines. This, along with the viral mutations that occur in eggs during the manufacturing process, results in a variable vaccine efficacy of between 40% and 60%.
Along with manufacturing-process
challenges, egg-based vaccines
are slow to produce and there would likely be an insufficient supply of eggs to produce the vaccines necessary in a pandemic setting.Our product concepts
Our influenza vaccine candidate is a multivalent vaccine consisting of mRNA encoding for two types of antigens, hemagglutinin (HA) and neuraminidase (NA), formulated in LNPs. We believe this combination of antigens has the potential to provide a protective immune response to influenza viruses.
Achievements to date and future milestones
We have formulation design and testing activities underway for our seasonal influenza mRNA vaccine program. We are testing prototype formulations in mice and ferret models, and plan to evaluate resulting data and consult with the FDA to select a candidate for
pre-clinical,
IND-enabling,
toxicology testing. Based on our current projected timeline, and if our preclinical studies are successful, we anticipate selecting a clinical candidate, in the first half of 2022 to undertakeIND-enabling
toxicology studies for a clinical Phase I trial starting in 2023 . We have an option to license LNP for our influenza vaccine candidate from an established LNP company. We will seek partnership with an established pharmaceutical company to conduct clinical development trials and, if our clinical studies are successful, commercialize our influenza vaccine.Gene therapies
There are thousands of genetic diseases caused by mutations in single genes. Patients with many of these diseases are not yet well-served by existing therapies. Although there are treatments for certain genetic diseases, sometimes the treatments alleviate symptoms temporarily or require organ, bone marrow, or stem cell transplants. These are costly, time-consuming, and logistically challenging. We aspire to develop our technology to edit the specifically targeted gene to treat such diseases by simple injections of mRNA/LNP formulations consisting of the gene and molecular machinery for its integration into the genome.
Our sickle cell disease gene product concept
Unmet need
Sickle cell disease affects about 100,000 people in just the United States and is prevalent in people of African and Middle Eastern descent. There is no cure for sickle cell disease, and current treatments focus on managing the pain crises and other effects such as anemia. Current treatment regimens—including blood transfusions and bone marrow transplants—are costly, invasive, and impractical for treating large segments of affected patient populations. Gene therapies currently in development for sickle cell disease are cell therapies, which require facilities close to the patient that can edit the cells outside of the body, posing an additional challenge for populations in remote areas or without adequate facilities to perform the editing.
Current approaches to gene therapy have challenges to overcome. Therapies that use adeno-associated viruses (AAVs) as vectors can encapsulate and deliver genetic material of up to 5,000 base pairs only, which limits the diseases to which this technology can be applied.
16
Product concept
Our RNA-based gene
product concept is to design a product candidate to deliver a healthy copy of the gene to stem cells. We believe our gene therapy concept has the potential to be:| • | Accessible: Based on our cost-competitive RNA platform and with an in vivo administration, we believe our therapy will enable us to bypass the need for facilities required to edit the cells ex vivo. |
| • | Targeted: The delivery technology targets specific cells in tissue. |
| • | One dose and done: Our strategy is to target precursor stem cells to provide long-lasting expression. |
| • | Versatile: Our therapy has the potential to encode for full-length genes and address genetic indications that require therapy in nondividing cells. |
Our work in gene therapy is supported by the Bill and Melinda Gates Foundation. This work involves reduction to practice of novel approaches for gene therapy using mRNA and cell/tissue targeting. We anticipate being ready for preclinical toxicology studies at the end of 2024.
Early Stage R&D
Our supra-seasonal influenza and antibody therapy targets utilize the mRNA platform technology used for our
SARSCoV-2
vaccine candidate. These targets are currently in the early stages of concept evaluation in terms of antigen or protein design andin-vitro
testing. If we succeed in these endeavors we anticipate selecting clinical candidates in 2024.GLOBAL RNA MANUFACTURING NETWORK
Our vision is to enable Africa, Asia, and Latin America to meet local demand through production outside the United States and Europe, from drug substance to product to fill and finish of mRNA vaccines and therapies, ideally in the country where the vaccine will be sold. If our vaccines and therapies are approved in these jurisdictions, we intend to contract with local manufacturers to produce our products, which we believe will enable the accessibility and cost competitiveness of our products.
If we obtain applicable regulatory approvals, we intend to create an interoperable network with local production facilities deploying our manufacturing process using modular design concepts that can be
constructed off-site and
set up more quickly than traditional construction models, so each facility will rely less on international supply chains to create vaccines and therapies for local needs.STRATEGIC COLLABORATIONS
Collaborators are part of our core strategy as we seek to accelerate our development of RNA therapies. We have relationships with research hospitals, universities, foundations, biotechnology companies, pharmaceutical companies, and nongovernmental organizations with expertise in our pipeline programs. During research and development stages, we seek collaborators to complement our preclinical studies and manufacturing capabilities. At the clinical development stage, we will seek established collaborators to codevelop or commercialize our product candidates. For vaccines, we are seeking companies with commercial capabilities that will receive rights to develop and commercialize our vaccine candidate. In this way, we can share the risk and reward of our portfolio while acquiring the capabilities required to launch commercial products. We seek partners aligned with our mission of making RNA accessible to the world.
Our decision to partner will be determined by the partner’s geographic scope and the complementary capabilities that partner can bring to support the commercialization of products. We have yet to choose either the
17
products for which we choose to partner, or the partners themselves; however, we may also choose to commercialize some early-stage programs without partners, as large-scale commercial capabilities may not be required for programs with a small patient population.
PLANT HEALTH PRODUCT PIPELINE
Overview
We plan to design, build, and sell a complete portfolio of products that growers can use throughout the food chain, from field to fork, to enhance, protect, and preserve produce and animals.
Our product pipeline is based on double-stranded RNA, or dsRNA, which works by regulating the expression of a carefully selected protein in the target organism, be it in plants, fungi, or animals (primarily insects or arachnids). This method can, with careful selection of the appropriate target, potentially be used to control a wide range of unwanted pests and problems.
An introduction to dsRNA and agriculture
As a tool for crop protection, dsRNA has several advantages. It is designed to impact the target pest and limit harm to, there is a long-established view that dietary intake of nucleic acids, including dsRNAs from plant viruses, does not present a health risk to humans and other vertebrates, and, as a result, the adoption of RNAi technology in agriculture is likely to present a lower human health risk than the use of conventional pesticides.
any non-targeted organisms.
Unlike many other pesticides, dsRNA degrades quickly in the environment, so it is typically undetectable after a few days, meaning in typical use, treated produce would contain low to no pesticide residue. Finally, in the event any residue remains, there is an established history of safe consumption of RNA molecules in human and animal food. According to a September 2020 report published by the Environmental Directorate of the Organization for Economic Cooperation and Development entitled Considerations for the Environmental Risk Assessment of the Application of Sprayed or Externally AppliedPesticides
ds-RNA-Based
Based on our toxicity testing and these advantages of dsRNA, GreenLight has requested a tolerance exemption from the EPA for the active ingredient contained in its first dsRNA product, GS2, which seeks to control Colorado Potato Beetle in potatoes and other solanaceous crops. If granted, such an exemption would be consistent with a category IV toxicity level, the EPA’s lowest level of pesticide toxicity under FIFRA.
Process for developing new products
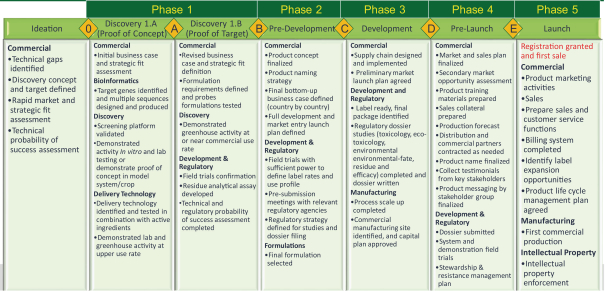
GreenLight uses a five-phase product development process for plant and animal health products as summarized in the following table. In general, in order for a product to move to a particular stage, it must successfully have met the requirements of the preceding stages.
18
Market opportunity
Given the versatility
of RNA-based solutions,
we believe that the markets for our products are large. In the near term, we intend to pursue more than $10 billion in addressable target markets for plant health, with the full launch of our first product anticipated in 2023.We define total addressable market as the global revenue opportunity available to pesticide solutions controlling a target pest or disease. In most instances, we do this by defining a relevant active ingredient market for the crop or crops where we intend to market our products and then making an assumption as to the percentage of that market that is spent on controlling the target pest or disease. We use data from AgbioInvestor and FAOSTAT (a database run by the Food and Agriculture Organization of the United Nations), and we use data purchased from third-party consultants to quantify the market and underpin assumptions.
We intend to develop products for our own distribution as well as for commercial partners. In doing so, we will focus our attention on the fresh fruits, vegetables, and nuts markets, which urgently need residue-free crop- protection products or have a strong association with the need to conserve honeybees. We will seek to serve the broadacre markets and international markets through partnerships with established multinational crop-protection companies and distributors. We intend to develop products that farmers trust and incorporate as a regular part of their annual crop-protection program.
Each of the initial products we are developing is intended to be specific to one target pest based on grower needs. We believe we can leverage our expertise in RNA in the future to target multiple pests as well as use our manufacturing platform and experience to make novel products at a cost that works for farmers.
What specific problems are we trying to solve?
Our Plant Health group is working to provide growers with highly effective tools to use within their normal cultural practices that avoid
disrupting non-target organisms
while leaving low to no residue in the treated produce. Today there are very few commercially available products that successfully combine these characteristics that growers, regulators, and consumers desire. Our primary focus for this mission is the successful deployment of carefully designed dsRNA. In order for our products to function successfully, the organism that needs to be managed must possess the appropriate cellular apparatus to process exogenous dsRNA to regulate protein biosynthesis. For these organisms, we intend to develop a portfolio of insecticides, acaricides, fungicides, and products that affect crop physiology and health, such as bio stimulants and herbicides.19
Insecticides and acaricides
Our insecticides and acaricides program is currently working on six major targets with a combined addressable target market size of $4.4 billion. These projects are distributed across various phases ranging from the most advanced, which is in
the pre-commercial phase
awaiting regulatory approval, to nascent candidates. We calculate addressable markets for our projects using market data from AgbioInvestor and FAOSTAT and information purchased from third-party consultants. We use this data as well as our industry knowledge to inform assumptions around expenditures to control the target pest or disease to arrive at the addressable market.Calantha, our program to control the Colorado potato beetle, has been submitted to the EPA for approval. Another, aimed at Varroa mites, is expected to be submitted to the EPA in 2022. We anticipate moving the program for diamond back moth to the field in 2022 or 2023, with EPA submission in 2023. Additionally, we project to submit our two spotted spider mite product to the EPA for approval in 2024.
Colorado potato beetle
Calantha, our product candidate for the Colorado potato beetle (), which decimates plants in the nightshade family and accounts for more than $500 million in crop loss annually, has gone from discovery to Environmental Protection Agency (EPA) submission in four years. The application is mixed with water and sprayed using standard agricultural practice over crops at a rate of 9.9 grams per hectare—less
Leptinotarsa decemlineata
than one-tenth the
rate at which many conventional industrial chemicals are normally used on fields. Consumption of the dsRNA, which itself degrades within days, causes the Colorado potato beetle to stop eating and expire from its own toxins while beneficial insects are unaffected. In the United States, we have tested this product over the last three annual growing seasons in Oregon, Washington, Wisconsin, New York, Maine and Idaho. We have also conducted field tests of the product in Spain, Germany and France.We believe the addressable market for protecting crops from the Colorado potato beetle is approximately $350 million. Assuming EPA approval in 2022, we anticipate full commercialization in 2023.
Widely recognized for its ability to develop resistance to pesticides, the Colorado potato beetle was first described as a pest in the United States in 1859.
We expect the price and performance of Calantha, the first-ever foliar RNA product—submitted for regulatory approval with the EPA in October 2020—to be competitive with other products currently available to farmers. We have conducted more than 100 field trials over four years to develop a product that is effective at just 9.9 g/hectare, an extremely low active ingredient use rate, equivalent to a spoonful of sugar spread on a football field.
Our testing has shown that Calantha is safe for honeybees, butterflies, and several
other non-target insects
and mammals at use rates 100 times higher than our recommended rate. It degrades in water and soil within three days to benign, natural nucleotides. The product works well with standard growers’ programs to control first- or second-generation Colorado potato beetle. It effectively controls all stages of the life of this beetle but is most effective on young larvae upto one-quarter inch
in length.In addition to being water soluble, this product contains additional inert ingredients to allow it to be mixed with other agricultural products and applied by farmers in a single spraying using common methods,
including low-water volume
(aerial or ground) or chemigation. Although conventional pesticides can require special protective equipment for farmers, we anticipate just basic work gloves will be required for this product.Varroa mites
Having acquired the rights to portions of Bayer’s topical RNA intellectual property portfolio, which
include bee-health assets,
we are developingan RNA-based syrup
that targets reproductive mites, is easy to use, and will add another tool in the limited Varroa-control market.20
We have been field testing our
RNA-based
product candidate for the Varroa destructor mite, which many beekeepers consider to be the top threat to honeybees and which has been detected in up to 90% of US hives, since March 2021 with the assistance of commercial beekeepers in Georgia, California, Florida, and Maine. To date, these tests demonstrate a measurable improvement in hive health. Hive health is measured by bee management personnel through a visual assessment of open broods (uncapped hive cells containing larvae) and closed broods (capped hive cells containing larvae) and the assignment of scores based on that assessment. The assessment also includes a scored evaluation of the overall health of the hive, including appearance and productivity of drones, brood health and queen health. Hive health measurements attempt to take into account a variety of factors other than mite count and include potential harmful effects from the pathogens mites can introduce into the hive, the potential harmful effects of chemical pesticides and other environmental factors that can affect overall hive health. Our tests measured hive health 12 weeks after application of our Varroa mite product and demonstrated a 20% improvement in open brood health (p=0.0193), a 20% improvement in closed brood health (p=0.0163) and a 17% improvement in overall hive health (p=0.0200), compared to hive health measured 12 weeks after application of the commercial standard chemical Varroa mite control product. As part of our hive health assessments, we also assess mite population and control before and after application of our product, using guidelines from the USDA’s Agricultural Research Services for measuring mite populations. Using these guidelines in our field tests, we observed a decline in mite population of 80% 6 weeks after treatment (p=0.0073) and 77% 12 weeks after treatment (p=0.0106) after the application of our product.About 3 million commercial honeybee colonies in the United States are used to pollinate more than 100 crops annually that are worth an estimated $15 billion, according to the U.S. Department of Agriculture. The parasitic Varroa mite reproduces in hives, feeds on honeybees, and spreads disease, destroying colonies across the globe. Now in the development phase, our product candidate targets the Varroa mite to protect bees, beekeepers, and pollination-dependent crops.
When we acquired rights from Bayer relating to its” for a discussion of several risks factors relating to our Varroa mite product. Nonetheless, until we complete our field trials, it is unclear whether we will be able to limit delivery of our product to bees and, through bees, to Varroa mites, in a manner that will effectively impede mite function.
bee-health
assets, Bayer disclosed to us that in laboratory tests its original Varroa mite product had been observed to have adverse effects on ladybugs. We are developing our own version of this product using our proprietary manufacturing process, and our product has a different composition than the Bayer product. We have observed adverse effects on ladybugs in laboratory tests of products made with our dsRNA manufacturing process but at 10x higher use rates than Bayer observed. We do not believe that such data will negatively impact our ability to secure a registration from the EPA or impact the attractiveness of our product to potential customers for two reasons. First, during the normal course of use, our product would be delivered in a sealed package directly to beehives, and it is atypical for ladybugs to enter a treated beehive during the proposed treating season. Accordingly, it is unlikely that the organisms that may be negatively affected would be exposed to the product during the normal course of use. Second, we believe that customers would conclude that the benefits of controlling Varroa mites outweigh the potential risks to ladybugs. See “Item 1A. Risk Factors
— Risks Related to our Animal Health Program
In preparation for seeking EPA approval of our Varroa mite product, we have been conducting laboratory and field tests, including a required high-dose test to assess risks associated with potential overexposure to the product. When we tested our Varroa mite product in the laboratory at the required level of ten times the field use rate, the higher concentration of the product caused the treated bee food to become highly viscous, which limited consumption and resulted in bee starvation. We did not observe these adverse effects either when our product was administered at the field use rate or when our product was administered at the high-dose rate in the field. Because our product is delivered in aformulation through a
ready-to-use
pre-measured
pouch delivery system, rather than through conventional spraying, we do not believe that our product presents a material risk that bees will be exposed to concentrations greater than the field use rate. For more information regarding potential adverse effects on regulatory approval of our Varroa mite product if the EPA does not agree to modify its safety factor protocol, see “Item 1A. Risk Factors
—
Risks Related to our Animal Health Program
—
The EPA will
21
evaluate our Varroa mite product without a precedent product, which may result in the need to conduct additional field trials and lengthen the regulatory review period.
If we cannot reduce bee mortality experienced in high-dose safety factor testing, the EPA may not approve our product or may impose labeling requirements that materially limit the commercial attractiveness of the product.
In order to submit a registration dossier to the EPA in 2022, we need to complete additional studies required for the initial submission, including a bee safety study that is only available seasonally. This study is part of the normal course of an EPA registration and generally cannot be conducted during cold winter months. We are currently conducting this study, and additional
non-target
organism studies are in progress during the spring of 2022 for inclusion in the dossier we plan to submit to the EPA. For the United States registration of our Varroa mite product, we will be able to submit for approval under the FIFRA regulations, and we expect to make the submission in the second or third quarter of 2022. In certain foreign jurisdictions, including the European Union, we expect that we will be required to apply for authorization of our Varroa mite product as an animal health product under applicable veterinary medicine regulations.Diamond Back Moth
The Diamond Back Moth () is sometimes called the cabbage moth because of its voracious appetite for consuming brassicas plants, which include cabbage, Brussels sprouts, and cauliflower, among others. It represents a global challenge to farmers and growers because its short life cycle allows it to rapidly develop resistance to existing crop protection products.
Plutella xylostella
By testing Diamond Back Moth larvae in greenhouse assays (where they are fed foliage treated with their specific RNA sequence combined with the different delivery technologies we have developed), we believe that the Diamond Back Moth can be controlled with a dsRNA-based pesticide. We are now testing delivery methods by which the product would be delivered to the field. If we successfully develop delivery mechanisms, we expect to enter the field-testing stage for the product. If a successful delivery mechanism is developed, we anticipate moving into
our pre-development phase
in 2022 and the development phase in 2023, with the goal of a 2026 launch, subject to receipt of regulatory approval.Two Spotted Spider Mite
Two spotted spider mites () (TSSM) are not insects but arachnids that feed on plants. All life cycle stages of the mite will cause damage to the plants upon which they feed. TSSM use their mouthparts to pierce cells on the surface of the leaf to suck out the contents, rendering the cell useless. TSSM will feed on a wide range of crops from Glasshouse ornamentals to tree nuts and fruits to corns and soybeans and can be found almost anywhere crops are grown. This project is currently in the discovery phase where we are seeing good control from
Tetranychus urticae
our on-plant assays
without any need to develop any specific delivery technology. We anticipate progressioninto pre-development in
2023 and move rapidly on to development in 2024 by having an initial focus on controlled environment crops.Fungicides
Our fungicides program currently has six major targets in the pipeline with a combined total addressable market of $7.2 billion. This includes botrytis, fusarium, powdery mildew, downy mildew, Asian soybean rust, and rice blast. We calculate addressable markets for our projects using market data from AgbioInvestor and FAOSTAT and information purchased from third-party consultants. We use this data as well as our industry knowledge to inform assumptions around expenditures to control the target pest or disease to arrive at addressable market.
Our fungicide programs for botrytis and powdery mildew control are currently being field tested, and we expect to make an EPA submission in 2023. We also expect to submit our fusarium program to the EPA in 2023.
22
Botrytis
Botrytis cinerea

We began testing our Botrytis product in California, New York and Italy in 2021 and anticipate one more year of field testing before applying to the EPA for product approval in 2023. Assuming an
18-24
month EPA approval cycle, we would expect to begin commercialization in 2025. California, a major market for this product, could lag EPA approval by up to a year or more. Because this product demonstrated disease control in the field on both of the crops that we tested (strawberries and grapes), we progressed the project into Phase 2, thepre-development
phase, at our portfolio review in December 2021. We believe we can move this project into Phase 3 of our development process at the end of 2022, which we believe would allow us to move through the remaining development phases and, subject to receipt of regulatory approval, into the market by 2025.Grapevine powdery mildew
Powdery mildew, caused by, is the most common and destructive disease affecting grapes. Mostly observed on the upper surface of leaves as a dusty gray or white coating, the disease also strikes the lower surface, young stems, buds, flowers, canes, and fruit. Severely infected leaves may exhibit mottling or deformity, including leaf curling and withering. Infected fruit turn grayish-white first, then exhibit a brown, rusted appearance and may crack, shrivel, or drop from clusters.
Erysiphe necator
We conducted our first season of field trials in 2021 in New York and California, and we were able to demonstrate disease control comparable to current leading chemical-control products. We anticipate one more year of field trials with the goal of regulatory submission in 2023. Assuming the EPA approves the product in 2025, we would expect to begin commercialization that year. California, a major market for this product, could lag EPA approval by up to a year or more, and
non-US
grape growing regions such as France could also take one or more additional years to obtain approval. Because this product demonstrated disease control in the field on both of the crops that we tested (strawberries and grapes), we progressed into Phase 2, thepre-development
phase, at our portfolio review in December 2021. We believe we can move this project into Phase 3 of our development process at the end of 2022, which we believe would allow us to move through the remaining development phases and, subject to receipt of regulatory approval, into the market by 2025.23
Fusarium Head Blight
Fusarium Head Blight is a disease of cereal crops most typically caused in the United States and Europe by, though some other Fusarium species are implicated. Fusarium species have a wide host range and can cause many different types of damage to crops depending upon the type of crop plant and its growth stage at the time of infection. In the form of the disease in which it infects the flowering ear of the cereal crop, Fusarium does not necessarily rob the farmer of yield but instead frequently produces mycotoxins as part of its metabolic process. These mycotoxins can cause serious illness and even death when consumed in small quantities (the primary mycotoxin is deoxynivalenol, or DON, known colloquially as vomitoxin), so there is a detection limit in process food stuffs of 11 ppm. dsRNA can be designed to inhibit the metabolic pathway that produces the mycotoxins. We believe the ability to do this is a key differentiator for GreenLight. The current fungicides available to the grower will control the pathogen but do not provide reliable suppression of the mycotoxins. This product is currently in the “Discovery 1B” phase described above, with GreenLight having demonstrated under controlled conditions that it can stop mycotoxin production on growing wheat. Based on our product development to date, we believe we can move this project into Phase 3 of our development process in 2022, which we believe would allow us to move through the remaining development phases and into the market by 2025.
Fusarium graminearum
Crop physiology
The market for crop physiology, crop health, and herbicides is approximately $25 billion, based on data compiled by AgbioInvestor. Our activities in these areas have taught us how to deliver dsRNA into cells, so we are expanding our research to give us further opportunities in the crop-protection market.
Limitations we are working to overcome
The use of dsRNA as a crop-protection technology has been proposed since the discovery of the mechanisms of RNAi in the 1990s. A key barrier to the development of dsRNA was the cost of manufacturing RNA itself.
Our proprietary cell-free technology aims to solve this problem. Other technical, commercial, and social challenges remain, with delivery as the next challenge. Not all organisms will readily uptake dsRNA in the way that the Colorado potato beetle and Varroa mite do, and we need to deploy strategies that overcome barriers for lepidoptera (the rate of breakdown in the gut) or plants (passage across membranes). Much of our
mid-
and long-term pipeline target work relates to addressing the challenge of extending environmental stability both within the gut of target insects and on the leaves of sprayed plants.Another perceived limitation of dsRNA is associated with its highly specific nature. While we believe that this specificity is a benefit because it can make the technology safer for beneficial insects and humans, we realize most products on the market are broad-spectrum, which is appealing to farmers because they enable farmers to control multiple pests or diseases at once.
Biotechnology and agriculture have a complicated history. We know that building trust with stakeholders is critical to ensuring smooth adoption. We seek to educate about dsRNA and the benefits of what it can do. By increasing awareness of the benefits of RNA biopesticides, we hope to form strong relationships with other sustainability-oriented initiatives and industry stakeholders who can help tell our story.
Our people and culture
At GreenLight, we celebrate the power of working together to address humanity’s challenges, meet the needs of underserved populations, and push the boundaries of scientific discovery. Our culture represents a team united by a common purpose of creating a more sustainable future by bringing food security, medicine, and
24
healthcare to everyone. From the very beginning, our founders believed that our way forward would be based on equality, diversity, and inclusion (ED&I). These founding principles guide us every day as we seek to identify, attract, retain, incentivize, and develop a highly talented workforce.
Qualified team
Building a platform through RNA manufacturing to address food and agriculture markets and human health markets in vaccines and gene therapies requires deep technical and scientific expertise.
As of March 21, 2022, we have 312 full-time employees. Within our workforce, 271 employees are engaged in research and development and manufacturing operations and 41 are engaged in the shared business-enabling functions. Approximately 53% of our team members who are focused on research and development have master’s degrees or higher and approximately 38% of our team members have PhDs. Our employees are not represented by any labor union nor any collective-bargaining arrangement with respect to their employment with us.
In addition to our regular workforce, we are grateful for the collaboration, contributions, and support of a network of industry advisors, consultants, contractors, and temporary staff who make up the overall GreenLight team.
With ED&I principles as the foundation, we are focused on cultivating a team with diverse backgrounds and perspectives. We consider how we can better serve our colleagues of different genders, ethnicities, generations, educational achievements, sexual orientations, workstyles, and more. Our current senior management team is diverse: 50% of our Executive team members and Senior Vice Presidents are female. Additionally, approximately 40% of our Executive team identifies as
a non-Caucasian racial
or ethnic group (Black or African American, Hispanic or Latino, American Indian or Alaska Native, Asian or Native Hawaiian, other Pacific Islander, or two or more races). Our management team is committed to continuing to build a diverse team and a culture of inclusion to ensure that diverse perspectives thrive.Numbers alone cannot capture the rich diversity of our company. However, we collect and report these numbers for transparency and as a marker of our continued efforts. Approximately 46% of our full-time employees self-identify as female and 45% of full-time employees self-identify as a
non-Caucasian
racial or ethnic group (as defined above). While we acknowledge that there is still work to be done, we are committed to doing our part to make real changes to address systemic bias and inequities.ENVIRONMENTAL, SOCIAL, AND GOVERNANCE (ESG) STRATEGY
Environmental and social impact is inherent to our purpose and the underlying reason our company was launched. We were founded to develop sustainable products for the biggest issues facing humanity and the planet. GreenLight scientists are developing new products for public health challenges and sustainable food production to feed a growing population. We believe our ESG strategy is fundamental to achieving our mission and underscores everything we do at GreenLight.
We are striving to optimize the environmental impact of our facilities and operations, but we recognize the greatest potential for impact is through our product development process and in our goal to design and
manufacture RNA-based products
to support human, animal and plant health more naturally and safely.Environmental
There is a need to take immediate action to address the environmental crisis that is forcing the reconsideration of how products are made, from our homes to our food, to our clothing. Many modern approaches to produce food and drugs to keep the growing population healthy have had a negative effect on the
25
health of the planet. Clear cutting forests for cattle, chemical residues on food, in the water and in the soil, nitrogen blooms in rivers, declining soil productivity, the loss of bees and other beneficial insects—these are all clear signs that the current system is not sustainable.
The world is running out of arable soil. For years, farmers have used effective petroleum-based chemical pesticides in the form of neonicotinoids, pyrethroids, carbamates, and organophosphates. Over time, these
non-targeted products
can have unintended negative consequences, including damage to beneficial insects and plants, and they can linger in the environment for years, eroding soil quality and polluting water resources.Using RNA, we can create targeted biocontrols for agriculture. Biology also offers a fundamental shift in how things are made and disposed of in a world where things grow and decay, creating circular, regenerative processes. Our goal is to have products that can help the environment, not harm it. GreenLight’s RNA is produced from materials using an enzymatic process and after application our RNA product candidates disappear in a few days. We believe that, because the active RNA ingredients in our product candidates quickly degrade in the environment, our product candidates will have the potential to be more sustainable, or greener, than traditional petroleum-based chemical pesticides.
We aim to provide farmerstargeted biocontrols that stop pests while protecting crops, honeybees, and land before and after harvest. If we help farmers create greener, cleaner crops, they can provide consumers with the greener, cleaner foods they demand. Additionally, we also intend to provide farmers with safer products to handle, while helping farming families promote more sustainable land for future generations.
with safe-to-use, cost-effective,
When we refer to a product or process as ‘green’ in the context of potential agricultural products, we are referring to the fact that dsRNA-based pesticides have the potential to leave little to no residue behind after use, resulting in a significant potential reduction in the toxins or other foreign matter released into local waterways, aquifers, or the food chain. Additionally, when we use the term ‘sustainable,’ we refer to our efforts to align economic development with environmental protection and human well-being as well as our anticipated obligations as a Public Benefit Corporation under § 362(a) of the Delaware General Corporation Law.
Social
Values and biases can be embedded in the technologies that are made, in the applications that are considered, and in the ways problems are addressed. Inclusion of those who have historically been left out of the development of new technologies is essential to building equitable and positive outcomes. GreenLight was born from a passion to make our world more sustainable and more equitable. Our vision is to enable Africa, Asia, and Latin America to meet local demand through local production. Our novel RNA manufacturing process—quick to start, built for scale, and using small bioreactors—may be part of the solution.
An ecosystem thrives with more diversity, and the inclusion of many different voices is essential to growing our company. Team members are empowered to bring their best ideas forward, and leaders are always open to listen and act. We challenge one another to discover breakthroughs that advance our science to deliver on a common cause: sustaining the planet, protecting our food, saving lives. With equity, diversity, and inclusion principles as the foundation, we are relentlessly focused on cultivating a team with diverse backgrounds and perspectives. We are always thinking about how we can better serve our colleagues of different genders, ethnicities, generations, educational achievement, sexual orientation, and workstyles. These values and initiatives are not just
a top-down corporate
statement; they are an intrinsic part of our culture.Governance
At GreenLight, we celebrate the power of working together to address humanity’s challenges, meet the needs of underserved populations, and push the boundaries of scientific discovery. Our culture represents a team
26
united by a common purpose of creating a more sustainable future by bringing food security, medicine, and healthcare to everyone. From the very beginning, our founders believed that our way forward would be based on equality, diversity, and inclusion (ED&I). These founding principles guide us every day as we identify, attract, retain, incentivize, and develop a highly talented workforce.
The following values are deeply coded within our business, mission, and culture:
| • | Care for everyone |
| • | Courage to achieve the impossible |
| • | Collaboration to propel our success |
| • | Commitment to science and doing the right thing, always |
Our culture is built on care, transparency, diversity, employee ownership and engagement, and a deep, humble respect for science. Transparency is essential to how we operate, to enable sharing of the insights and tools that enable our platform to grow, as well as to build trust and accountability with all our stakeholders.
We have selected independent directors and scientific advisory board members with decades of experience. Our board of directors and management team will leverage that experience and consider the interests of stockholders, customers, employees, suppliers, academic researchers, governments, communities, and other stakeholders to pursue long-term value for our company and drive the sustained health of our global community.
27
COMPETITION
We are aware of only one large company, Bayer AG, that has human health and agricultural capabilities similar to our company. Other competitors split into either human health or agricultural market categories.
Human health
Our competitors are biotechnology companies working on indications similar to our pipeline, mRNA companies, large pharmaceutical companies, and academia.
We are aware of several large pharmaceutical and biotechnology companies, as well as smaller, early-stage companies, pursuing the development of products and disease indications we are targeting. These include major vaccine and therapeutics companies such as Roche Holding AG, AbbVie, GlaxoSmithKline (GSK), Merck & Co Inc, Sanofi, Pfizer, AstraZeneca, Johnson & Johnson, and Novavax.
Among RNA specialist companies, BioNTech and Moderna already have
COVID-19
vaccines on the market, while CureVac N.V., Arcturus Therapeutics Inc., Translate Bio, Daiichi Sankyo, Elixirgen Therapeutics, and Providence Therapeutics have clinical trials underway. Specialized therapeutics companies such as Alnylam Pharmaceuticals, Editas Medicine, and Dicerna Pharmaceuticals also compete against GreenLight.Agriculture
We believe that our technology platform coupled with our research and development expertise and commercial strategy set us apart from others in the food and agricultural market. Because crop protection is a mature industry, there are several companies targeting similar insects and fungi and investing in effective products. These include larger companies such as Syngenta and Bayer, as well as smaller companies such as Provivi, Vestaron, and Biotalys. Creating the sustainable food system we know is possible will require the expertise and dedication of many people bringing many new products to market. We look forward to collaborating with many companies, even those we have called out as competitors, to achieve that future.
Platform
Our platform, and our ability to manufacture biological molecules, is a key competitive advantage and driver of future growth. Ginkgo Bioworks, Zymergen, and Codexis, among others, have
sophisticated know-how and
may emerge as competitors.INTELLECTUAL PROPERTY
We strive to protect and enhance proprietary technology, inventions, and improvements that are commercially important to the development of our business, including seeking, maintaining, and defending patent rights, whether developed internally or licensed from third parties. We also rely on trade secrets to develop, strengthen, and maintain proprietary positions that may be important for the development of our business. We additionally may rely on regulatory protection afforded through data exclusivity, market exclusivity, and patent-term extensions, where available.
Our commercial success may depend in part on our ability to: obtain and maintain patent and other proprietary protection for commercially important technology, inventions
and know-how related
to our business; defend and enforce our patents and patent applications; preserve the confidentiality of our trade secrets; maintain and defend our trademark registrations and applications; and operate without infringing the valid, enforceable patents and other intellectual property and proprietary rights of third parties. Our ability to limit third parties from making, using, selling, offering to sell, or importing our products or using our proprietary methods may28
depend on the extent to which we have rights under valid and enforceable licenses, patents, trademarks or trade secrets that cover these activities. We cannot be sure that patents will be granted with respect to any of our pending patent applications or with respect to any patent applications filed by us in the future, nor can we be sure that any of our existing patents or any patents that may be granted to us in the future will be commercially useful in protecting our commercial products or methods of manufacturing and using the same or that they will prevent others from commercializing competing products or technology.
Patents
As of March 22, 2022, we had approximately 40 patent families (the term “patent family” is used here to denote patents and applications claiming priority to a common patent application) in various fields of our business. Of those patent families, approximately seven families relate to RNA production; approximately six families relate to other human health-related technologies; approximately 17 families relate to crop protection and bee health; approximately three families relate to production of sugars; and approximately seven families relate to process control and compound production. The number of families may change if we file additional applications or obtain additional issued patents or if we abandon any of our pending or issued patents. We continue to evaluate the costs and potential benefits of patent protection in various jurisdictions. In connection with such evaluations, we may abandon pending applications or issued patents.
Individual patent terms extend for varying periods of time, depending on the date of filing of the patent application, the date of patent issuance, and the legal term of patents in the countries in which they are obtained. In most countries in which patent applications are filed, including the United States, the patent term is 20 years from the date of filing of thefrom country to country, and depends on many factors, including the type of patent, the scope of its coverage, the availability of legal remedies in a particular country, and the validity and enforceability of the patent.
first non-provisional application
to which priority is claimed. Under certain circumstances, a patent term can be extended. For example, in the United States, a patent’s term may be lengthened by patent term-adjustment, which compensates a patentee for administrative delays by the U.S. Patent and Trademark Office in reviewing and granting a patent; by patent-term extension for certain patents covering products requiring regulatory approval prior to being sold or methods of using or making such products; or may be shortened if a patent is terminally disclaimed over an earlier-filed patent. However, the actual protection afforded by a patent varies ona product-by-product basis,
Our seven RNA production patent families include four families directed to RNA production platforms. All four families, including all of the issued patents in such families, contain claims directed to methods of manufacture of RNA and/or related processes. One such platform family includes U.S. Patent No. 10,858,385, the issued claims of which protect certain aspects of our process for production of dsRNA. This family also includes a pending U.S. continuation and a number of foreign applications pending in Argentina, Australia, Brazil, Canada, Chile, China, Costa Rica, the European Patent Office, Japan, Hong Kong, India, Indonesia, Israel, Malaysia, Mexico, New Zealand, Russia, Singapore, South Africa, South Korea, Thailand, and Ukraine. The projected expiration for U.S. Patent No. 10,858,385 is in 2037, not including any term adjustments or extensions if applicable.
Another RNA production platform family contains U.S. Patent No. 10,954,541 along with a pending U.S. continuation, issued patents in India and Indonesia, and a number of foreign applications pending in jurisdictions including Australia, Brazil, Canada, Chile, China, Costa Rica, the European Patent Office, Hong Kong, Israel, Japan, Malaysia, Mexico, New Zealand, South Korea, Singapore, Thailand, and Ukraine. The projected expiration for U.S. Patent No. 10,954,541 is in 2037, not including any term adjustments or extensions if applicable.
The third RNA production platform family contains U.S. Patent No. 11,274,284, along with a pending U.S. continuation, issued patents in China, Indonesia, Japan, and Singapore, and additional foreign applications pending in jurisdictions including the European Patent Office, Israel, India and South Korea. The projected
29
expiration date for U.S. Patent No. 11,274,284 is in 2036, not including any term adjustments or extensions if applicable.
The fourth RNA production platform family relates to methods for production of mRNA that may have applicability to our next generation approach for such production. This family contains a United States application along with applications recently filed in a number of foreign jurisdictions, including, Australia, Canada, China, the European Patent Office, India, Israel, Japan, Korea, Malaysia, Singapore, and South Africa. All applications in this family are national stage applications from International Application No. PCT/US2020/025824. If the U.S. patent application in this family were allowed, the projected expiration of the resultant patent would be in 2040, not including any term adjustments or extensions if applicable.
The RNA production families also contain patent applications directed to various improved compositions and processes. These include, for example, two families, each consisting of an international patent application related to plasmid templates for production of RNA products, proteins and enzymes of interest. If we were to file U.S. national stage patent applications from these international applications and any claims were to be allowed, they would have projected expiration dates in 2040 and 2041, not including any term adjustments or extensions if applicable.
Other Human Health Patent Families
We currently have six additional Human Health specific patent families. One family includes International Application No. PCT/US2021/042015 and a related U.S. patent application directed to compositions and methods of treatment related to our ongoing research in the field of gene therapy. If the U.S. patent application were to be allowed, the projected expiration of the resultant patent would be in 2041, not including any term adjustments or extensions if applicable. Two families each contain a pending United States provisional patent application related to the same research. Another of the families contains a U.S. provisional application directed to mRNA compositions, including the company’s proposed COVID vaccine.
Bee Health and Crop Protection Families
Our 17 Bee Health and Crop Protection patent families include seven Bee Health patent families and ten Crop Protection patent families.
Bee Health Patent Families
Four of our Bee Health patent families contain claims directed to or related to our proposed varroa mite product and/or its use. The first such family.” This family consists of U.S. Patent Nos. 8,962,584, 9,662,348, and 10,801,028, which contain composition and method of treatment claims expected to expire in 2030, not including any term adjustments or extensions if applicable. This family further includes issued patents in China, France, Germany, Israel, Italy, Mexico, New Zealand, Russia, South Africa, Turkey, the United Kingdom, and Ukraine and pending patent applications in Canada, Chile, China, and Mexico.
is co-owned with
Yissum and subject to an exclusive license to us of Yissum’s ownership interest in commercial rights to this patent family. For more information on this license, see “Item 1. Business—Intellectual Property Agreements — Bayer Acquisition Agreement
Another such Bee Health patent family
is co-owned with
the United States Department of Agriculture. This family consist of U.S. Patents Nos. 10,100,306, 10,927,374, and 9,540,642 with composition and method of treatment claims expected to expire in 2034, not including any term adjustments or extensions if applicable. The family further includes a pending U.S. continuation; issued patents in Australia, Israel, Russia, Ukraine, and South Africa; and pending applications in Australia, Argentina, Canada, Chile, China, European Patent Office, India, Mexico, New Zealand, and Uruguay.30
The third such Bee Health patent family includes U.S. Patent No. 10,907,152, which has composition and method of treatment claims and is expected to expire in 2036, not including any term adjustments or extensions if applicable. This family further comprises a pending U.S. continuation; foreign patents in China, Israel, and South Africa; and pending foreign applications in jurisdictions including Argentina, Australia, Brazil, Canada, Chile, European Patent Office, India, Mexico, New Zealand, Russia, Ukraine, Uruguay.
The fourth such Bee Health patent family contains a pending U.S. application, Serial No. 17/013,330 along with pending applications in Australia and New Zealand. If claims of the U.S. application were to be allowed, the resultant patent would have a projected expiration in 2040, not including any term adjustments or extensions if applicable.
Crop Protection Patent Families
We have ten Crop Protection patent families. Three such families relate to nucleic acid compositions for control of Colorado potato beetle. One of those three families includes U.S. Patent No. 11,142,768 (expected to expire in 2039, not including any term adjustments or extensions if applicable) with composition claims directed to Calantha, our proposed Colorado potato beetle product, along with two pending related U.S. applications, and pending foreign patent applications in Australia, Brazil, Canada, China, European Patent Office, India, Japan, New Zealand, Russia, and Ukraine. Another of those three Colorado potato beetle patent families contains U.S. Patent No. 11,185,079, which has composition claims and an expected expiration in 2039 (not including any term adjustments or extensions if applicable), one pending related U.S. application, and pending foreign applications in Australia, Brazil, Canada, China, European Patent Office, India, Japan, New Zealand, Russia, and Ukraine. The third such family has one pending U.S. patent application.
Our seven other Crop Protection patent families include: one family with a pending international patent application directed to compositions for controlling lepidopteran pests; one family with a pending U.S. patent application and an international patent application directed to compositions for control of a fungus; three provisional applications directed to compositions for control of lepidopteran pests; a provisional application directed to compositions for controlling fungi; and one provisional application directed to compositions for improved product stability.
Sugar Platform Patent Families
We have three patent families related to enzymatic production of sugars. One family consists of issued U.S. Patent Nos. 10,316,342, 10,577,635, and 10,704,067, all of which contain method of production claims and are expected to expire in 2038, not including any term adjustments or extensions if applicable. That family also consists of a pending U.S. continuation and foreign applications in Australia, Brazil, Canada, Chile, China, Colombia, European Patent Office, Hong Kong, India, Indonesia, Japan, Mexico, Russia, South Korea, and Thailand.
A second sugar platform family comprises United States and pending foreign applications in a number of jurisdictions recently filed as national stage entries of International Application No. PCT/US2019/067113. If the U.S. patent application in this family were to be allowed, the resultant patent claims would have a projected expiration in 2039, not including any patent term adjustments or extensions if applicable.
Our third family consists of a pending provisional patent application directed to improved compositions useful for production of sugars.
Trade secrets
GreenLight’s technology-related intellectual property that are not patent-protected are maintained as confidential information and trade secrets. We employ a variety of safeguards to protect our confidential information and trade secrets, including contractual arrangements that impose obligations of confidentiality and security, digital security measures, and physical security precautions.
31
With respect to contractual arrangements, we protect our confidential and proprietary information by requiring our employees to execute nondisclosure and assignment of invention agreements upon commencement of their employment. Agreements with our employees also bar them from using the proprietary rights of third parties in the course of their employment or disclosing to us any confidential information of third parties.
We require confidentiality and material transfer agreements from third parties that receive our confidential data or materials, and we also incorporate confidentiality and material transfer precautions into our research and collaboration agreements.
We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems
Trademark and domain names
GreenLight owns 4 U.S. trademark applications relating to the GreenLight name and logo and the Calantha brand in the United States and other jurisdictions around the world. We are still evaluating whether we want to release some or all of our products under the GreenLight brand, or whether we want to develop new brands applicable to specific product pipelines. We also have a registered domain name for our website found at www.greenlightbiosciences.com.
Intellectual Property Agreements
Bayer Acquisition Agreement
We entered into an Assignment and License Agreement with and Bayer CropScience LLP (“
Bayer
”) dated December 10, 2020 (the “Bayer Acquisition Agreement
”), pursuant to which we acquired from Bayer certain intellectual property rights related to (i) RNA technology used to control Varroa mites, Nosema, and bee viruses, which includes assignments of patents and a license to research, develop and sell methods and products in such field with the use of Bayer“know-how”
with respect to such technology, and the assignment of Bayer’s rights under a license agreement with Yissum Research and Development Company of the Hebrew University of Jerusalem LTD (the “Yissum License
”), and (ii) technology used to control the Colorado potato beetle and canola flea beetle, including a license to research, develop and sell methods and products in such field with the use of Bayer patents and“know-how”
with respect to such technology. Under the Bayer Acquisition Agreement, we were obligated to make a closing payment to Bayer equal to $2,000,000 as well as certain milestone payments equal to up to $2,000,000 in the aggregate, in the event that certain regulatory approvals are achieved with respect to the aforementioned technologies.We also agreed to indemnify Bayer against losses arising out of GreenLight’s recklessness, willful misconduct, violation of law or breaches of representations or warranties under the Bayer Acquisition Agreement or activities related to the use of intellectual property assigned to GreenLight thereunder. The Bayer Acquisition Agreement shall survive for so long as the assigned patents remain in effect; provided, that the parties do not terminate the Bayer Acquisition Agreement earlier in accordance with its terms.
Under the Bayer Acquisition Agreement, GreenLight was assigned Bayer’s rights and obligations under the Yissum License. Pursuant to the Yissum License, we were granted an exclusive, worldwide license to make use of the relevant technology to develop, manufacture, market, distribute, or sell covered products, including an exclusive, worldwide license under Yissum’s interest in any bee health patents jointly owned with GreenLight. Notwithstanding the exclusive license, Yissum retains the right to practice the jointly-owned patents in ways that will not result in competition with GreenLight, including the right of the Hebrew University of Israel (the “research organizations to do the same, provided that no such license directly or indirectly harms our commercial interest in the relevant patents and products.
University
”) to practice the inventions for the University’s own internal research and educational purposes, and to license other academic andnot-for-profit
32
We also have the right, but not the obligation, to prosecute in our own name and at our own expense any infringement of patents jointly owned with Yissum, but in making our decision whether to assert infringement, we must give consideration to the views of Yissum. In order to settle any such infringement suit, we must obtain the consent of Yissum.
Pursuant to the Yissum License, we agreed to pay Yissum a running royalty percentage in the low single digits on net sales of the licensed products. The License ends on abasis upon the later of the date of expiration of the last valid licensed patent, the end of regulatory exclusivity for a product, or 20 years from the date of first sale.
country-by-country
Pursuant to the Yissum License, we are liable for any loss, injury or damage whatsoever caused to our employees or to any person acting on our behalf or to the employees of Yissum or to any person acting on our behalf or to any third party, by reason of GreenLight’s acts or omissions pursuant to the Yissum License or by reason of any use made by GreenLight of the licensed technology and products. Moreover, we will compensate, indemnify, defend, and hold harmless Yissum or any person acting on its behalf or any of its employees or the University or representatives of the University against any liability imposed upon them by GreenLight’s acts or omissions or which derive from GreenLight’s use, development, manufacture, marketing, sale or sublicensing of any product or licensed technology unless it has been determined by an adjudicator of last resort that that the particular damage, loss or expense was caused by a particular indemnitee’s gross negligence or willful misconduct.
Either party may terminate the Yissum License upon written notice in the event of a bankruptcy or similar proceeding of the other party. Yissum may terminate the license agreement if we do not commercialize products within a reasonable timeframe, with certain exceptions, if it provides written notice and we do not cure such failure within a certain timeframe; or if we have an uncured lapse in necessary insurance coverage; or if we unreasonably fail to respond to third-party claims against the patents or technology licensed under the Yissum License.
Patent expiration is a legal determination under the laws of each relevant jurisdiction worldwide. Third parties may review public patent filings and make their own determination as to patent expiration based on the available documents. The last to expire U.S. Patent under the Yissum License is expected to expire in 2031.
Bill & Melinda Gates Foundation
We entered into a Grant Agreement with the Bill and Melinda Gates Foundation dated July 20, 2020, as amended by that certain Amendment 1 to Grant Agreement dated May 25, 2021 (as amended, the “
Gates Grant
”) pursuant to which we were awarded a grant in the amount of $3,343,151, payable in milestone tranches, for research regarding treatment and curative therapies for sickle cell disease and/or durable suppression of HIV in developing countries. We utilize the Gates Grant funds to explore new, low cost capabilities for the in vivo functional cure of sickle cell disease as well as the durable suppression of HIV. In the event that GreenLight has materially breached the Grant Agreement, the Foundation may demand repayment of the Gates Grant funds.Acuitas Therapeutics
Development & Option Agreement
In August 2020, we and Acuitas entered into a development and option agreement, or the Acuitas Option Agreement. Under the Acuitas Option Agreement, the parties agreed to jointly develop certain products combining our RNA constructs with Acuitas’s LNPs. Each party granted the other party a worldwide,
non-exclusive,
royalty-free license under its proprietary technology to conduct the joint research. We pay Acuitas’s personnel costs and external expenses incurred in performing research in accordance with a work plan under the Acuitas Option Agreement. Under the Acuitas Option Agreement, Acuitas granted us options to33
obtain non-exclusive, worldwide,
sublicensable licenses under Acuitas’s patent rightsand know-how related
to LNP technology, or Acuitas LNP Technology, with respect to three specified targets, or Reserved Targets, to develop and commercialize one or more therapeutic products incorporating Acuitas LNP Technology and our RNA constructs. We paid Acuitas a technology access fee of $750,000 at the outset of the Option Agreement. Thereafter, we are obligated to pay an annual technology maintenance fee of $250,000 for each option that has not been exercised and target reservation and maintenance fees of $100,000 per Reserved Target until such Reserved Target is removed from the Reserved Target list or until we exercise an option with respect to such Reserved Target.On exercise of the first option, we were required to pay a $1.5 million option exercise fee after execution of the first
non-exclusive
license. On exercise of the second and third options, we are required to pay a $1.75 million and $2.75 million option exercise fee after execution of the second andthird non-exclusive licenses,
respectively.Unless earlier terminated, the Acuitas Option Agreement will remain in effect until the first to occur of (1) all options are exercised, and (2) three years from the effective date, except that we can choose to extend the three-year term for an additional two years. Either party may terminate the Acuitas Option Agreement for an uncured material breach of the other party or upon the other party’s bankruptcy or a similar event. We may terminate the Acuitas Option Agreement at our convenience following written notice to Acuitas. To GreenLight’s knowledge, the last to expire U.S. patent under the Acuitas Option Agreement will expire in 2041 if the last filed relevant U.S. patent application currently identified by Acuitas is allowed. However, additional intellectual property, including patents, may still be added to the Acuitas Option Agreement or may not be known to us. Therefore the last to expire patent under that Option Agreement may change. Moreover, patent expiration is a legal determination under the laws of each relevant jurisdiction worldwide. Third parties may review public patent filings and make their own determination as to patent expiration based on the available documents.
Any jointly developed intellectual property under the Acuitas Option Agreement is jointly owned by the parties in an
undivided one-half interest
to such joint intellectual property.Non-Exclusive License
AgreementIn January 2021, we exercised the first option under the Acuitas Option Agreement and entered intoRoyalty Term. Either party may terminate the Acuitas License Agreement for an uncured material breach of the other party or upon the other party’s bankruptcy or a similar event. We may terminate the Acuitas License Agreement at our convenience following written notice to Acuitas.
a non-exclusive license
agreement with Acuitas, or the Acuitas License Agreement. Acuitas granted usa non-exclusive, worldwide,
sublicensable license under the Acuitas LNP Technology to research, develop, manufacture, and commercially exploit a vaccine product consisting of our RNA constructs and Acuitas’s LNPs. We paid Acuitas an option exercise fee of $1.5 million. Under the Acuitas License Agreement, we are required to pay Acuitas an annual license maintenance fee of $1 million until we achieve a particular development milestone. Acuitas is entitled to receive potential clinical, regulatory, and commercial milestone payments of up to $17.25 million in the aggregate. With respect to the sale of each licensed product by us, our affiliates or our sublicensees, Acuitas is entitled to receive low single digit percentage royalties on net sales of the licensed product in a given country until the last to occur, in such country, of (i) the expiration of all licensed patent rights covering the licensed product, (ii) expiration of any regulatory exclusivity for the licensed product, or (iii) ten years from the first commercial sale of the licensed product, or Royalty Terms. We are entitled to certain royalty reductions and offsets with respect to each licensed product in a given country if no licensed patents cover the licensed product or if we are required to obtain rights to third party patents that relate to LNP technology. Unless earlier terminated, the Acuitas License Agreement will remain in effect until the expiration of thelast-to-expire
Additional intellectual property, including patents, may still be added to the Acuitas License Agreement or may not be known to us. Therefore the last to expire patent under that License Agreement may change.
34
Moreover, patent expiration is a legal determination under the laws of each relevant jurisdiction worldwide. Third parties may review public patent filings and make their own determination as to patent expiration based on the available documents. To GreenLight’s knowledge the last to expire U.S. patent under the Acuitas License Agreement will expire in 2041 if the last filed relevant U.S. patent application currently identified by Acuitas is allowed.
GOVERNMENT REGULATION
We are using our RNA manufacturing platform to develop products for human health and agriculture, and we are subject to laws and regulations for those markets. These regulations currently apply to development and testing of our products and in the future will apply to manufacturing, import, export, marketing, and sale of products.
Human health products
We are developing human health products and gene therapies that include vaccines
for COVID-19,
shingles, influenza, and sickle cell anemia. In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act (“FDCA
”) and biologics under the Public Health Service Act (“PHSA
”). Both drugs and biologics also are subject to other federal, state, and local statutes and regulations. The FDA and other regulatory authorities at federal, state, and local levels, as well as in foreign countries, extensively regulate, among other things, the research, development, testing, manufacture, quality control, import, export, safety, effectiveness, labeling, packaging, storage, distribution, record keeping, approval, advertising, promotion, marketing, post-approval monitoring, and post-approval reporting of drugs and biologics. We, along with third-party contractors, will be required to navigate the various preclinical, clinical and commercial approval requirements of the governing regulatory agencies of the countries in which we wish to conduct studies or seek approval or licensure of our product candidates. The process of obtaining regulatory approvals and the subsequent compliance with applicable federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources.U.S. biologics regulation
In the United States, biological products such as gene therapies and vaccines are subject to regulation under the Federal Food, Drug, and Cosmetic Act, the Public Health Service Act, and other federal, state, local and foreign statutes and regulations. The process required by the FDA before biologics may be marketed in the United States generally involves the following:
| • | completion of preclinical laboratory tests and animal studies performed in accordance with the FDA’s Good Laboratory Practice requirements (“ GLPs ”); |
| • | submission to the FDA of an investigational new drug application (“ IND ”), which must become effective before clinical trials may begin; |
| • | approval by an institutional review board (“ IRB ”) or ethics committee at each clinical site before the trial is commenced; |
| • | performance of adequate and well-controlled human clinical trials to establish the safety, purity and potency of the proposed biologic product candidate for its intended purpose; |
| • | preparation of and submission to the FDA of a biologics license application (“ BLA ”), after completion of all pivotal clinical trials; |
| • | satisfactory completion of an FDA Advisory Committee review, if applicable; |
| • | a determination by the FDA within 60 days of its receipt of a BLA to file the application for review; |
35
| • | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities at which the proposed product is produced to assess compliance with cGMP, and to assure that the facilities, methods and controls are adequate to preserve the biological product’s continued safety, purity and potency, and of selected clinical investigation sites to assess compliance with Good Clinical Practices (“GCPs ”); and |
| • | FDA review and approval of the BLA to permit commercial marketing of the product for particular indications for use in the United States. |
Prior to beginning the first clinical trial with a product candidate in the United States, we must submit an IND to the FDA. An IND is a request for authorization from the FDA to administer an investigational new drug to humans. The central focus of an IND submission is on the general investigational plan and the protocol(s) for clinical studies. The IND also includes results of animal and studies assessing the toxicology, pharmacokinetics, pharmacology, and pharmacodynamic characteristics of the product; chemistry, manufacturing, and controls information; and any available human data or literature to support the use of the investigational product. An IND must become effective before human clinical trials may begin. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within
in vitro
the 30-day time
period, raises safety concerns or questions about the proposed clinical trial. In such a case, the IND may be placed on clinical hold and the IND sponsor and the FDA must resolve any outstanding concerns or questions before the clinical trial can begin. Submission of an IND therefore may or may not result in FDA authorization to begin a clinical trial.Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators in accordance with GCPs, which include the requirement that all research subjects provide their informed consent for their participation in any clinical study. Clinical trials are conducted under protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A separate submission to the existing IND must be made for each successive clinical trial conducted during product development and for any subsequent protocol amendments. Furthermore, an independent IRB for each site proposing to conduct the clinical trial must typically review and approve the plan for any clinical trial and its informed consent form before the clinical trial begins at that site, and must monitor the study until completed. Regulatory authorities, the IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk or that the trial is unlikely to meet its stated objectives. Some studies also include oversight by an independent group of qualified experts organized by the clinical study sponsor, known as a data safety monitoring board, which provides authorization for whether or not a study may move forward at designated check points based on access to certain data from the study and may halt the clinical trial if it determines that there is an unacceptable safety risk for subjects or other grounds, such as no demonstration of efficacy. There are also requirements governing the reporting of ongoing clinical studies and clinical study results to public registries.
For purposes of BLA approval, human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| • | Phase 1—The investigational product is initially introduced into healthy human subjects or patients with the target disease or condition. These studies are designed to test the safety, dosage tolerance, absorption, metabolism and distribution of the investigational product in humans, the side effects associated with increasing doses, and, if possible, to gain early evidence on effectiveness. |
| • | Phase 2—The investigational product is administered to a limited patient population with a specified disease or condition to evaluate the preliminary efficacy, optimal dosages and dosing schedule and to identify possible adverse side effects and safety risks. Multiple Phase 2 clinical trials may be conducted to obtain information prior to beginning larger and more expensive Phase 3 clinical trials. |
36
| • | Phase 3—The investigational product is administered to an expanded patient population to further evaluate dosage, to provide statistically significant evidence of clinical efficacy and to further test for safety, generally at multiple geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit ratio of the investigational product and to provide an adequate basis for product approval. |
In some cases, the FDA may require, or companies may voluntarily pursue, additional clinical trials after a product is approved to gain more information about the product.
These so-called Phase
4 studies may also be made a condition to approval of the BLA. Concurrent with clinical trials, companies may complete additional animal studies and develop additional information about the biological characteristics of the product candidate, and must finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, must develop methods for testing the identity, strength, quality and purity of the final product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.BLA submission and review by the FDA
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, the results of product development, nonclinical studies and clinical trials are submitted to the FDA as part of a BLA requesting approval to market the product for one or more indications. The BLA must include all relevant data available from preclinical and clinical studies, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls, and proposed labeling, among other things. Data can come from company-sponsored clinical studies intended to test the safety and effectiveness of a use of the product, or from a number of alternative sources, including studies initiated by independent investigators. The submission of a BLA requires payment of a substantial application user fee to the FDA, unless a waiver or exemption applies.
Within 60 days following submission of the application, the FDA reviews a BLA submitted to determine if it is substantially complete before the FDA accepts it for filing. The FDA may refuse to file any BLA that it deems incomplete or not properly reviewable at the time of submission and may request additional information. In this event, the BLA must be resubmitted with the additional information. Once a BLA has been accepted for filing, the FDA’s goal is to review standard applications within ten months after the filing date, or, if the application qualifies for priority review, six months after the FDA accepts the application for filing. In both standard and priority reviews, the review process may also be extended by FDA requests for additional information or clarification. The FDA reviews a BLA to determine, among other things, whether a product is safe, pure and potent and the facility in which it is manufactured, processed, packed or held meets standards designed to assure the product’s continued safety, purity and potency. The FDA may also convene an advisory committee to provide clinical insight on application review questions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving a BLA, the FDA will typically inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP and are adequate to assure consistent production of the product within required specifications. Additionally, before approving a BLA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP. If the FDA determines that the application, manufacturing process or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional
37
information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
After the FDA evaluates a BLA and conducts inspections of manufacturing facilities where the investigational product and/or its drug substance will be produced, the FDA may issue an approval letter or a Complete Response Letter (CRL). An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications. A CRL will describe all of the deficiencies that the FDA has identified in the BLA, except that where the FDA determines that the data supporting the application are inadequate to support approval, the FDA may issue the CRL without first conducting required inspections, testing submitted product lots, and/or reviewing proposed labeling. In issuing the CRL, the FDA may recommend actions that the applicant might take to place the BLA in condition for approval, including requests for additional information or clarification. The FDA may delay or refuse approval of a BLA if applicable regulatory criteria are not satisfied, require additional testing or information and/or require post-marketing testing and surveillance to monitor safety or efficacy of a product.
If regulatory approval of a product is granted, such approval will be granted for particular indications and may entail limitations on the indicated uses for which such product may be marketed. For example, the FDA may approve the BLA with a Risk Evaluation and Mitigation Strategy (REMS) to ensure the benefits of the product outweigh its risks. A REMS is a safety strategy implemented to manage a known or potential serious risk associated with a product and to enable patients to have continued access to such medicines by managing their safe use, and could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA also may condition approval on, among other things, changes to proposed labeling or the development of adequate controls and specifications. Once approved, the FDA may withdraw the product approval if compliance
with pre- and
post-marketing requirements is not maintained or if problems occur after the product reaches the marketplace. The FDA may require one or more Phase 4 post-market studies and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization, and may limit further marketing of the product based on the results of these post-marketing studies.Expedited development and review programs
The FDA offers a number of expedited development and review programs for qualifying product candidates. For example, the fast track program is intended to expedite or facilitate the process for reviewing product candidates that are intended to treat a serious or life-threatening disease or condition and demonstrate the potential to address unmet medical needs for the disease or condition. Fast track designation applies to the combination of the product candidate and the specific indication for which it is being studied. The sponsor of a fast track product candidate has opportunities for more frequent interactions with the applicable FDA review team during product development and, once a BLA is submitted, the product candidate may be eligible for priority review. A fast track product candidate may also be eligible for rolling review, where the FDA may consider for review sections of the BLA on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the BLA, the FDA agrees to accept sections of the BLA and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the BLA.
A product candidate intended to treat a serious or life-threatening disease or condition may also be eligible for breakthrough therapy designation to expedite its development and review. A product candidate can receive breakthrough therapy designation if preliminary clinical evidence indicates that the product candidate, alone or in combination with one or more other drugs or biologics, may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. The designation includes all of the fast track program features, as well as more intensive FDA interaction and guidance beginning as early as Phase 1 and an organizational commitment to expedite the development and review of the product candidate, including involvement of senior managers.
38
Any marketing application for a drug or biologic submitted to the FDA for approval, including a product candidate with a fast track designation and/or breakthrough therapy designation, may be eligible for other types of FDA programs intended to expedite the FDA review and approval process, such as priority review and accelerated approval. A BLA is eligible for priority review if the product candidate is designed to treat a serious or life-threatening disease or condition, and if approved, would provide a significant improvement in safety or effectiveness compared to available alternatives for such disease or condition. For original BLAs, priority review designation means the FDA’s goal is to take action on the marketing application within six months of
the 60-day filing
date (as compared to ten months under standard review).Additionally, product candidates studied for their safety and effectiveness in treating serious or life-threatening diseases or conditions may receive accelerated approval upon a determination that the product candidate has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. As a condition of accelerated approval, the FDA will generally require the sponsor to perform adequate and well-controlled post-marketing clinical studies to verify and describe the anticipated effect on irreversible morbidity or mortality or other clinical benefit. Products receiving accelerated approval may be subject to expedited withdrawal procedures if the sponsor fails to conduct the required post-marketing studies or if such studies fail to verify the predicted clinical benefit. In addition, the FDA currently requires as a condition for accelerated
approval pre-approval of
promotional materials, which could adversely impact the timing of the commercial launch of the product.Fast track designation, breakthrough therapy designation, priority review, and accelerated approval do not change the standards for approval but may expedite the development or approval process. Even if a product candidate qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or decide that the time-period for FDA review or approval will not be shortened.
Post-approval requirements
Biologics are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to record-keeping, reporting of adverse experiences, periodic reporting, product sampling and distribution, and advertising and promotion of the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims, are subject to prior FDA review and approval. There also are continuing, annual program fees for any marketed products. Biologic manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP, which impose certain procedural and documentation requirements. Changes to the manufacturing process are strictly regulated, and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting requirements. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance.
The FDA may withdraw approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical studies to assess new safety risks; or imposition of distribution restrictions or other restrictions under a REMS program. Other potential consequences include, among other things:
| • | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
39
| • | fines, warning letters, or untitled letters; |
| • | clinical holds on clinical studies; |
| • | refusal of the FDA to approve pending applications or supplements to approved applications, or suspension or revocation of product license approvals; |
| • | product seizure or detention, or refusal to permit the import or export of products; |
| • | consent decrees, corporate integrity agreements, debarment or exclusion from federal healthcare programs; |
| • | mandated modification of promotional materials and labeling and the issuance of corrective information; |
| • | the issuance of safety alerts, Dear Healthcare Provider letters, press releases and other communications containing warnings or other safety information about the product; or |
| • | injunctions or the imposition of civil or criminal penalties. |
The FDA closely regulates the marketing, labeling, advertising and promotion of biologics. A company can make only those claims relating to safety and efficacy, purity and potency that are approved by the FDA and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion
of off-label uses.
Failure to comply with these requirements can result in, among other things, adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties. Physicians may prescribe legally available products for uses that are not described in the product’s labeling and that differ from those tested and approved by the FDA.Such off-label uses
are common across medical specialties. Physicians may believe thatsuch off-label uses
are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, restrict manufacturer’s communications on the subjectof off-label use
of their products.Biosimilars and reference product exclusivity
The Affordable Care Act, signed into law in 2010, includes a subtitle called the Biologics Price Competition and Innovation Act, or BPCIA, which created an abbreviated approval pathway for biological products that are biosimilar to or interchangeable with
an FDA-licensed reference
biological product. The FDA has issued several guidance documents outlining an approach to review and approval of biosimilars.Biosimilarity, which requires that there be no clinically meaningful differences between the biological product and the reference product in terms of safety, purity, and potency, can be shown through analytical studies, animal studies, and a clinical study or studies. Interchangeability requires that a product is biosimilar to the reference product and the product must demonstrate that it can be expected to produce the same clinical results as the reference product in any given patient and, for products that are administered multiple times to an individual, the biologic and the reference biologic may be alternated or switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic. However, complexities associated with the larger, and often more complex, structures of biological products, as well as the processes by which such products are manufactured, pose significant hurdles to implementation of the abbreviated approval pathway that are still being worked out by the FDA.
Under the BPCIA, an application for a biosimilar product may not be submitted to the FDA until four years following the date that the reference product was first licensed by the FDA. In addition, the approval of a biosimilar product may not be made effective by the FDA until 12 years from the date on which the reference product was first licensed. During
this 12-year period
of exclusivity, another company may still market a competing version of the reference product if the FDA approves a full BLA for the competing product containing that applicant’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the40
safety, purity and potency of its product. The BPCIA also created certain exclusivity periods for biosimilars approved as interchangeable products. At this juncture, it is unclear whether products deemed “interchangeable” by the FDA will, in fact, be readily substituted by pharmacies, which are governed by state pharmacy law.
A biological product can also obtain pediatric market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods and patent terms.
This six-month exclusivity,
which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of a pediatric study in accordance withan FDA-issued “Written
Request” for such a study.Laboratory licensing and certification requirements
We are planning to partner with contract laboratories who are subject to the Clinical Laboratory Improvement Amendments of 1988 (“
CLIA
”), which requires all clinical laboratories to meet certain quality-assurance, quality-control, and personnel standards.Agricultural products
We are developing insecticides and fungicides to protect crops and acaricides to protect honeybees that are beneficial to crops. In the United States, the development, testing, and commercialization of these products are regulated by the EPA through the Federal Food, Drug, and Cosmetic Act (“
FFDCA
”), the Food Quality Protection Act (“FQPA
”) and the Federal Insecticide, Fungicide and Rodenticide Act (“FIFRA
”).In general, FIFRA prohibits the sale or distribution of any pesticide, a product category that includes the insecticides, fungicides, and acaricides we are developing, unless that pesticide is registered with the EPA. To register a pesticide with the EPA, the applicant must demonstrate that the product will not cause unreasonable adverse effects on human health or the environment. These adverse effects include any unreasonable risk to man or the environment, taking into account the economic, social, and environmental costs and benefits of the use of the pesticide, as well as any human dietary risk from residues that result from use of the pesticide in or on any food consistent with the FFDCA. In the course of its evaluation of a pesticide, EPA assesses the impact that a pesticide may have on endangered species and
non-target
organisms.Because our products contain novel
RNA-based
active ingredients, there will generally be no previously registered pesticide product containing that active ingredient and, as a result, the use of each of our products will require a new registration under FIFRA and the establishment of a tolerance under Section 408 of the FFDCA or the issuance of a tolerance exemption.In order for the EPA to register a pesticide:
| • | the applicant must first conduct specified studies to evaluate mammalian toxicology, toxicological effects to non-target organisms in the environment (ecotoxicological exposures), and the product’s physical and chemical properties; |
| • | the applicant must then submit to the EPA a registration dossier that includes data demonstrating that the product does not pose unreasonable risks; |
| • | the EPA will conduct both scientific and administrative reviews of the dossier, including a thorough evaluation of submitted safety data and completion of risk assessments for human dietary and ecotoxicological exposures; |
| • | if the EPA identifies any risks that appear to exceed regulatory standards or any other deficiencies in the dossier, it will ordinarily issue a letter identifying the deficiencies; |
| • | the applicant will have one or more opportunities to address any deficiencies, including the submission of factors that mitigate any risks identified in the EPA’s risk assessments; this process may involve ongoing submissions and coordination with the EPA to address any unresolved concerns; and |
41
| • | the EPA will undertake various stages of internal review prior to making a final decision on the application. |
The Pesticide Registration Improvement Act, enacted in 1996 and subsequently renewed, can serve to reduce the data requirements and timeline related to regulatory approvals for biopesticides when compared to other pesticides, with EPA approvals typically received within 16 to 24 months, compared with 36 months or longer for conventional chemical pesticides.
As part of the pesticide registration process, the EPA under its FFDCA authority establishes tolerances for pesticide chemicals, which consist of limits on pesticide residues that may remain on feed or feed commodities. In some cases, the EPA may issue a tolerance exemption when the chemical will have no impact on human health.
Even if a FIFRA registration is granted, the EPA has the authority to revoke the registration or impose limitations on the use of any of our pest management products if we do not comply with the regulatory requirements, if unexpected problems occur with a product, or if the EPA receives other newly discovered adverse information.
In addition to the approval by the EPA, we are required to obtain regulatory approval from the appropriate regulatory authorities in individual states and foreign jurisdictions before we can market or sell any pest management product in those jurisdictions. In most U.S. states, local authorizations typically take one to three months after EPA approval. In other states, such as California, Arizona and New York, regulatory authorities require additional data specific to their respective jurisdictions, and the process for having a product approved or denied can last an additional two to three months, or longer, for these states.
Outside the United States, the registration process varies by jurisdiction and can take between 24 and 84 months to complete. In most instances, initial submissions to foreign regulatory authorities will not occur until after a U.S. registration has been secured. Moreover, foreign governments typically require up to two seasons of locally generated field efficacy data on crop/pest combinations before a product dossier can be submitted for review. For example, in the EU, we would need to obtain authorization under Regulation (EC) No 1107/2009, which sets forth rules for the authorization, sale, use, and control of plant protection products, in order to market our products, and regulators may seek to require our products to comply with maximum residue levels under Regulation (EC) NO 396/2005.
In some instances, California and Canada will conduct joint reviews with the EPA, which allows some pesticides to receive concurrent approvals in California, Canada and the United States. California and foreign jurisdictions also require us to submit product efficacy data. Historically, the EPA has not required the submission of product efficacy data, but may request it.
The microbial strains used in our agricultural manufacturing process are also regulated by the EPA under the Toxic Substances Control Act (“
TSCA
”). In some circumstances, TSCA requires entities to provide notice to the EPA prior to the manufacture or importation of new microorganisms, called a Microbial Commercial Activity Notice (“MCAN
”). Persons intending to manufacture or import these microorganisms for commercial purposes in the United States must submit an MCAN to the EPA at least 90 days before such manufacture or importation. The EPA has 90 days to review the submission in order to determine whether the microorganism may present an unreasonable risk to human health or the environment. If the EPA makes that determination, the EPA may impose appropriate regulatory restrictions on the microorganism.Finally, a number of our products may require registration or approval under various state regulatory programs, including those relating to fertilizers, auxiliary plant substances, soil amendments, beneficial substances and/or biostimulants.
42
ITEM 1A. Risk Factors
Investing in New GreenLight Common Stock involves a high degree of risk. Before you decide to invest in any of our securities, you should consider carefully the risks described below, together with the other information contained in this annual report, including our consolidated financial statements and the related notes appearing at the end of this annual report. We believe the risks described below are the risks that are material as of the date of this annual report. If any of the following risks actually occur, our business, results of operations and financial condition would likely be materially and adversely affected. In these circumstances, the market price of New GreenLight Common Stock could decline, and you may lose part or all of your investment. This annual report also contains forward-looking statements that involve risks and uncertainties. See “Cautionary Note Regarding Forward-Looking Statements.” Our actual results could differ materially and adversely from those anticipated in these forward-looking statements as a result of certain factors, including, but not limited to, those set forth below.
Risks Relating to Our Business and Industry
We are an early-stage biotechnology company without any products or services currently available for sale and we may not be able to successfully develop or bring products or services to market.
In our human health program, we have
five pre-IND product
candidates, while in our plant health program, we have seven product candidates we hope to bring to market by 2027; however, there is no assurance that our future operations will generate any revenue. If we cannot develop a marketable product or generate sufficient revenues, we may be required to suspend or cease operations.We have not generated any product revenues to date and expect to incur losses and negative cash flow for the foreseeable future.
We have generated substantial accumulated losses since inception. Our net losses were $28.7 million, $53.3 million and $112.3 million for the years ended December 31, 2019, 2020 and 2021, respectively. As of December 31, 2021, we had an accumulated deficit of $253.6 million. We will need to generate significant revenues to achieve profitability, and we may not be able to achieve and maintain profitability in the near future. We have derived substantially all of our revenues through grants and research partnerships with third parties and we are unable to accurately predict whether these sources of revenue will be available to us in the future. Our future success will depend on our ability to bring products to market for the first time and grow consistent revenue associated with those products. The research, testing and regulatory pathways for each of the products in our product pipeline are complex and we can offer no assurance that we will bring the products in our pipeline to market, that any of those products will be profitable or that we will generate overall profit or positive cash flow in the future. The net losses we incur may fluctuate significantlythatof our results of operations may not be a good indication of our future performance. These fluctuations may cause our stock to be volatile compared to other stocks in the market.
from year-to-year and quarter-to-quarter, such
a period-to-period comparison
We will require substantial additional funds to complete our research and development activities, and, if additional funds are not available, we may need to significantly scale back or cease our business.
Between our founding and December 31, 2021, we raised net proceeds of $218.8 million from the sale of preferred stock, and from 2020 to 2021 we raised $67.0 million from the issuance of debt and convertible notes (including approximately $13.5 million of the PIPE Prepayment), which we have dedicated to the development of our RNA platform, human health product pipelines and plant health product pipelines. As of December 31, 2021, we held approximately $31.4 million in cash and cash equivalents (including approximately $13.5 million of the PIPE Prepayment). On February 2, 2022, we consummated the Business Combination and raised gross proceeds of approximately $136.4 million, which included funds held in ENVI’s trust account (after giving effect to redemptions of $194.9 million) and proceeds from the PIPE Financing of $124.3 million (inclusive of the PIPE Prepayment), before deducting estimated transaction expenses of $25.0 million. We have invested and will continue to invest in property, plant and equipment, and human capital and will require substantial funds to bring
43
the current products in our product pipeline to market and to grow our business by researching, developing, and protecting products not currently in our product pipeline. Our current available funds are not sufficient for all of these activities and, as a result, there is substantial doubt about our ability to continue as a going concern.
Based on our history of losses, we do not expect that we will be able to fund our longer-term capital and liquidity needs through our current cash balances and operating cash flow alone. To fund our longer-term capital and liquidity needs, we will need to secure additional capital. The amount of capital we will need will be subject to change depending on, among other things, the success of our efforts to grow revenue and our efforts to continue to effectively manage expenses.
When we raise additional funds through future issuances of equity or convertible debt securities, our existing stockholders could suffer significant dilution, and any new equity securities we issue could have rights, preferences and privileges superior to those of holders of our common stock. Any debt financing that we may secure in the future could involve restrictive covenants relating to our capital raising activities and other financial and operational matters, which may make it more difficult for us to obtain additional capital and to pursue business opportunities, including potential acquisitions. We may not be able to obtain additional financing on terms favorable to us, if at all. If we are unable to obtain adequate financing or financing on terms satisfactory to us when we require it, our ability to continue to support our business growth and to respond to business challenges and opportunities could be significantly impaired, and our business may be adversely affected.
Our financing needs may also increase substantially depending on the results of our research, trials and development for products and costs arising from additional regulatory approvals. We may not succeed in raising additional funds in a timely manner. The timing of our need for additional funds will depend on a number of factors, which are difficult to predict or may be outside of our control, including:
| • | the resources, time and costs required to initiate and complete our research and development and to initiate and complete studies and trials and to obtain regulatory approvals for additions to our product pipeline; |
| • | progress in our research and development programs; |
| • | the timing and amount of milestone, royalty and other payments; and |
| • | costs necessary to protect any intellectual property rights. |
If our estimates and predictions relating to any of these factors are incorrect, we may need to modify our business plans.
Our ability to raise funds will depend upon many factors, including conditions in the debt and equity capital markets, as well as investor perception of our creditworthiness and prospects. If we are unable to raise funds on acceptable terms, we may not be able to execute our business plan, take advantage of future opportunities, or respond to competitive pressures or unanticipated requirements. This may seriously harm our business, financial condition and results of operations. If we are not able to continue operations, investors may suffer a complete loss of their investments in our securities.
Our strategy assumes that we will collaborate with larger companies to develop and commercialize the products in our pipeline and if those collaborations are not successful or available to us at all, we may not be able to successfully commercialize our products.
We are seeking and will continue to seek collaboration arrangements with third parties for the development or commercialization of our products. These arrangements are complex and time-consuming to negotiate, document, implement and maintain and, as a small company, we may not have the same bargaining power as the larger companies we seek to collaborate with and the terms of any collaborations or other arrangements that we may establish may not be favorable to us.
44
Due to our limited resources and access to capital, we must make decisions on the allocation of resources to certain programs and product candidates; these decisions may prove to be wrong and may adversely affect our business.
We have limited financial and human resources and intend to initially focus on research programs and product candidates for a limited set of indications. As a result, we may forgo or delay pursuit of opportunities with other product candidates or for other indications that later prove to have greater commercial potential or a greater likelihood of success.
It is difficult to predict the time and cost of development of our pipeline products, which are produced by or based on a relatively novel and complex technology and are subject to many risks, any of which could prevent or delay revenue growth and adversely impact our market acceptance, business and results of operations. We may also determine not to pursue, or change the timing or order of, our product candidates.
Our company has product candidates with very complex and different sales and marketing channels, the development of which will put significant burdens on us and which we may not be able to develop as effectively as competitors.
We will have very different sales and marketing channels if the products in our pipeline are to reach customers in their respective markets, requiring us to develop distinct sales, marketing, and distribution methods. In particular, the human, agricultural and plant health markets have different customers and distribution channels. Building, managing and maintaining such a sales and marketing infrastructure will require us to hire experts in the field, implement complex systems, establish collaborations with third parties effectively across various geographies and understand disparate regulatory regimes. Our ability to effectively engage in these steps is untested, making it impossible for us to accurately predict the level of success we will achieve.
We have a limited operating history and funding, which may make it difficult to evaluate our product development, product prospects and overall likelihood of success.
We commenced operations in 2008. We have a limited operating history as a company, which makes it difficult to predict future operations. The product candidates and the markets we hope to serve have shifted since we commenced operations and as such, our operational experience with our current product pipeline and target markets is shorter than the full period of our operations. We have been operating our plant health product pipelines since 2016 and our human health product pipelines since 2019. Our approach to the discovery and development of product candidates had not been validated by the commercial introduction of products and we cannot guarantee that the products currently in our product pipeline, or any other products or services, will have future commercial value. Our programs will require substantial additional development and research, both in time and resources, before we are in a position to receive regulatory approvals and begin generating revenue in connection with the sale of product candidates. We have not yet demonstrated the ability to complete a large-scale, pivotal clinical trial, obtain regulatory approval for any product, manufacture a product at commercial scale, or arrange for a third party to do so on our behalf, or conduct sales and marketing activities necessary for successful product commercialization. Consequently, predictions about our future success or viability may not be as accurate as they could be if we had a longer operating history or a history of successfully developing and commercializing products.
All of our products require rigorous, time-consuming and expensive regulatory examination and approval before they can be commercialized and some or all of our products may not receive this approval.
Any products that we are currently developing or may develop in the future will be subject to extensive governmental regulations relating to development, trials, manufacturing and commercialization. Rigorous studies, clinical trials and extensive regulatory licensure and approval processes are required to be successfully completed in the United States and in many foreign jurisdictions, such as South Africa, the European Union and Japan, before a new product may be offered and sold in any of these countries or regions. Satisfaction of these and other regulatory requirements is costly, time-consuming, uncertain and subject to unanticipated delays.
45
Studies and trials for regulatory licensure and approval are expensive and difficult to design and implement, in part because they are subject to rigorous regulatory requirements. Because any product that we develop in the future will be based on new technologies, we expect that it will require extensive research and development and necessitate substantial manufacturing and processing costs. In addition, costs to treat potential side effects that may result from a product we develop may be significant.
Please refer to risk factor sections on our human health and animal health products for more regulatory risk information.
We have yet to establish sales, marketing or distribution capabilities, and if we are unable to establish these capabilities, we may not be successful in commercializing our product candidates if they are approved.
We have not yet established a sales, marketing or product distribution infrastructure for our product candidates, which are still in various stages of development. To achieve commercial success for any product for which we obtain marketing approval, we will need to establish a sales and marketing organization within the United States and develop a strategy for sales outside of the United States. In addition, as we begin to commercialize our products, we will need to hire, develop, train personnel with expertise in marketing and selling products in each of those markets.
Our growth strategy requires us to introduce new products, in addition to those in our current pipeline, that achieve market acceptance.
In order to reach our growth objectives, we must introduce new products in addition to our current pipeline of product candidates, and future new products. Research, development and regulatory approval for our products involve risks of failure inherent in the development of novel and complex products. These risks include the possibility that:
| • | our products may not perform as expected; |
| • | we may be unable to capitalize on successful innovation because we may choose not to incur the expense of patenting our discoveries in all jurisdictions or may be unable to obtain patents in the jurisdictions in which we wish to obtain patents; |
| • | any strategy of discovering additional vertical markets beyond plant, animal and human health for the use of RNA may be infeasible, limiting our growth; |
| • | our products may not receive necessary regulatory permits and governmental clearances in the markets in which we intend to sell them; |
| • | our competitors may develop new products or improve existing products that may make our products uncompetitive; |
| • | the lower cost of RNA produced by us may not translate equally or at all into lower prices for the products that use it; |
| • | our products may be difficult to produce on a large scale; |
| • | intellectual property and other proprietary rights of third parties may prevent us or our collaborators from making, marketing or selling our products; |
| • | we or our collaborators may be unable to fully develop or commercialize products in a timely manner or at all; and |
| • | third parties may develop superior or equivalent products. |
Accordingly, if we experience any significant delays in the development or introduction of new products or if our new products do not achieve market acceptance, our business, operating results and financial condition would be adversely affected.
46
We will need to expand our organization, and we may experience difficulties in managing this growth, which could disrupt operations.
As of December 31, 2021, we had 285 full-time employees, an increase in headcount of over 146% since December 31, 2019. We expect we will continue to experience significant growth in the number of our employees and the scope of our operations, particularly in the areas of product candidate development, regulatory affairs and manufacturing. We draw our talent from geographic and subject matter markets where the demand for the skills we seek are in the highest demand of any global labor market and we may have difficulty identifying, hiring and integrating and retaining new personnel. Future growth would impose significant additional responsibilities on our management, including the need to identify, recruit, maintain, motivate, integrate and retain additional employees, consultants and contractors. Also, our management may need to divert a disproportionate amount of our attention away fromand devote a substantial amount of time to managing these growth activities. We may not be able to effectively manage the expansion of our operations, which may result in weaknesses in our infrastructure, give rise to operational mistakes, loss of business opportunities, loss of employees and reduced productivity among remaining employees. Our expected growth could require significant capital expenditures and may divert financial resources from other projects, such as the development of product candidates. If our management is unable to effectively manage our growth, our expenses may increase more than expected, our ability to generate and/or grow revenues could be reduced, and we may not be able to implement our business strategy. Our future financial performance and our ability to commercialize our product candidates and compete effectively will depend, in part, on our ability to effectively manage any future growth.
our day-to-day activities
Many of the companies that we compete against for qualified personnel and consultants have greater financial and other resources, different risk profiles, more established brands and a longer history in the industry than we do. If we are unable to continue to attract and retain high-quality personnel and consultants, the rate and success at which we can discover and develop product candidates and operate our business will be limited.
We
use RNA-based molecular
biology triggers for many of the products in our product pipeline and the successful commercialization of these products will depend on public perceptionsof RNA-based products.
The successful commercialization of our product candidates depends, in part, on public acceptance of modern biotechnology techniques and the use of RNA to regulate the expression of genes and production of proteins in human health and agricultural products. Negative public perceptions about RNA and molecular regulation of gene expression can also affect the regulatory environment in the jurisdictions in which we are targeting the sale of our products and the commercialization of our product candidates. Any increase in such negative perceptions or any restrictive government regulations in response
to RNA-based products
could have a negative effect on our business and may delay or impair the sale of our products or the development or commercialization of our product candidates. Public pressure may lead to increased regulation and legislation for products produced using biotechnology and this could adversely affect our ability to sell our product or commercialize our product candidates.Our accounting predecessor, GreenLight, has identified material weaknesses in its internal controls of financial reporting. If we are unable to remediate the material weaknesses, or if we identify additional material weaknesses or otherwise fail to maintain effective internal control over financial reporting, this may result in material misstatements or restatements of our consolidated financial statements or cause us to fail to meet our periodic reporting obligations.
As a public company, New GreenLight is required to maintain internal control over financial reporting. Management may be unable to effectively and timely implement controls and procedures that adequately respond to the increased regulatory compliance and reporting requirements of a public company. If we are unable to comply with the requirement to maintain internal control over financial reporting, we may be unable to maintain compliance with securities law requirements regarding timely filing of periodic reports or applicable stock
47
exchange listing requirements, investors may lose confidence in our financial reporting, and our stock price may decline as a result.
Three material weaknesses have been identified in GreenLight’s internal control over financial reporting as of December 31, 2021. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis. The three material weaknesses identified in GreenLight’s internal controls result from:
| • | The failure to maintain a sufficient complement of personnel in its accounting and reporting department to ensure adequate segregation of duties such that appropriate review and monitoring of its financial records are executed. |
| • | The failure to design and implement adequate information systems controls, including access and change management controls. |
| • | The failure to maintain a sufficient complement of accounting and financial reporting resources commensurate with GreenLight’s financial reporting requirements. |
The first two material weaknesses were identified in connection with the preparation and audit of GreenLight’s financial statements as of December 31, 2020 (which audit was completed in September 2021), and had not been remediated as of December 31, 2021.
We have begun implementation of a plan to remediate these material weaknesses. These remediation measures are ongoing and include hiring additional accounting and financial reporting personnel and selecting and implementing new systems for our financial and enterprise resource planning. These remediation measures may be time-consuming and costly, and there is no assurance that they will ultimately have the intended effects.
If we identify any new material weaknesses or significant deficiencies in the future, any such newly identified material weakness or significant deficiency could limit our ability to prevent or detect a material misstatement of our annual or interim financial statements.
Moreover, our independent registered public accounting firm is not required to attest to the effectiveness of our internal control over financial reporting until after we are no longer an “emerging growth company” as defined in the JOBS Act. At that time, our independent registered public accounting firm may issue an adverse report if it is not satisfied with the level at which our internal control over financial reporting is documented, designed, or operating.
Under applicable employment laws, we may not be able to enforce covenants not to compete.
Our employment agreements generally include covenants not to compete. These agreements prohibit our employees, if they cease working for us, from competing directly with us or working for our competitors for a limited period. We may be unable to enforce these agreements under the laws of the jurisdictions in which our employees work.
Our business may be affected by litigation and government investigations.
We may from time to time receive inquiries and subpoenas and other types of information requests from government authorities and others and we may become subject to claims and other actions related to our business activities. While the ultimate outcome of investigations, inquiries, information requests and legal proceedings is difficult to predict, defense of litigation claims can be expensive, time-consuming and distracting, and adverse resolutions or settlements of those matters may result in, among other things, modification of our business practices, costs and significant payments, any of which could have a material adverse effect on our business, financial condition, results of operations and prospects.
48
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
Many of our employees, including our senior management, were previously employed at other biotechnology or pharmaceutical companies, including our potential competitors. Some of these employees may have executed proprietaryin connection with such previous employment. Although we try to ensure that our employees do not use the proprietary information
rights, non-disclosure and non-competition agreements
or know-how of
others in their work for us, we may be subject to claims that we or these employees have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such employee’s former employer. We are not aware of any threatened or pending claims related to these matters or concerning the agreements with our senior management, but future litigation may be necessary to defend against such claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management.We are exposed to a risk of substantial loss due to claims that may be filed against us in the future because our insurance policies may not fully cover the risk of loss associated with our operations.
We are exposed to the risk of having claims seeking monetary damages being filed against us for loss or harm suffered by participants of our preclinical and clinical studies or for loss or harm suffered by users of any products that may receive approval for commercialization in the future, or in connection with loss or harm from any other product, for example, agricultural products, that may be tested or receive approval for commercialization in the future. In either event, the FDA, EPA or the regulatory authorities of other countries or regions may commence investigations of the safety and effectiveness of any such trial or commercialized product, the manufacturing processes and facilities or marketing programs utilized in respect of any such trial or products, which may result in mandatory or voluntary recalls of any commercialized product or other significant enforcement action such as limiting the indications for which any such product may be used, or suspension or withdrawal of approval for any such product. Similar risks exist in connection with the testing, use, or sale of any product we may develop or commercialize. If our products are used for an application they are not intended for, become adulterated or mislabeled we may need to recall such products. A widespread product recall could result in significant losses due to the costs of a recall, the destruction of product inventory, and lost sales due to the unavailability of product for a period of time. We could also suffer losses from a significant product liability judgment against us. A significant product recall or product liability case could also result in adverse publicity, damage to our reputation, and a loss of confidence in our products, which could have an adverse effect on our business,
Significant disruptions of information technology systems or security breaches could adversely affect our operations.
We are increasingly dependent upon information technology systems, infrastructure and data to operate our business ourselves and on vendors who operate aspects of our technology infrastructure for us. In the ordinary course of business, we collect, store and transmit large amounts of confidential information (including, among other things, trade secrets or other intellectual property, proprietary business information and personal information). It is critical that we do so in a secure manner to maintain the confidentiality and integrity of such confidential information.
Attacks on information technology systems are increasing in their frequency, levels of persistence, sophistication and intensity, and they are being conducted by increasingly sophisticated and organized groups that include state actors, criminal organizations and individuals who can bring significant resources and expertise to bear. Public reports indicate that state actors have specifically targeted companies developing
COVID-19
vaccines with the intent of stealing trade secrets or disabling information technology systems associated with vaccine development and we may be unable to defend against these state actors who have significantly more resources at their disposal than we do.49
Our information technology systems, and those of third-party vendors with whom we contract are also vulnerable to service interruptions, security breaches from inadvertent or intentional actions by our employees, third-party vendors, and/or business partners, or from cyber-attacks by malicious third parties. Cyber-attacks could include the deployment of harmful malware,social engineering and other means to affect service reliability, and could threaten the confidentiality, integrity, and availability of information. For example, we recently experienced a ransomware attack that briefly interrupted access to two of our servers. Although in this instance we were able to rely our backup systems to restore access promptly, without making a ransom payment and without loss of data, our defenses against future cyber-attacks may not be successful.
ransomware, denial-of-service attacks,
Significant disruptions of our information technology systems, or those of our third-party vendors, or security breaches could adversely affect our business operations and/or result in the loss, adulteration, misappropriation and/or unauthorized access, use or disclosure of, or the prevention of access to, confidential information, including, among other things, trade secrets or other intellectual property, proprietary business information and personal information, and could result in financial, legal, business, and reputational harm to us.
Any failure or perceived failure by us or any third-party collaborators, service providers, contractors or consultants to comply with our privacy, confidentiality, data security or similar obligations to third parties, or any data security incidents or other security breaches that result in the unauthorized access, release or transfer of sensitive information, including personally identifiable information, may result in governmental investigations, enforcement actions, regulatory fines, litigation or public statements against us, could cause third parties to lose trust in us or could result in claims by third parties asserting that we have breached our privacy, confidentiality, data security, or similar obligations, any of which could have a material adverse effect on our reputation, business, financial condition, or results of operations. Moreover, data security incidents and other security breaches can be difficult to detect, and any delay in identifying them may lead to increased harm. While we have implemented data security measures intended to protect our information technology systems and infrastructure, there can be no assurance that such measures will successfully prevent service interruptions or data security incidents.
We are subject to anti-corruption, anti-bribery and anti-money laundering laws and regulations in various jurisdictions around the world which are subject to rigorous enforcement, and we can face serious consequences for violations.
We expect
our non-U.S. activities
to increase over time and to include countries that have more prevalent corruption than found in the U.S., increasing our exposure to anti-corruption, anti-bribery and anti-money laundering laws and regulations. These include the U.S. Foreign Corrupt Practices Act of 1977, and as amended, the U.S. domestic bribery statute contained in 18 U.S.C. § 201, the U.S. Travel Act, the U.K. Bribery Act and possibly other anti-corruption, anti-bribery and anti-money laundering laws and regulations in the jurisdictions in which we do business, both domestic and abroad. The FCPA and other anti-corruption laws generally prohibit companies, their employees, agents, representatives, business partners and third-party intermediaries from corruptly promising, authorizing, offering, or providing, directly or indirectly, anything of value to government officials, political parties, or candidates for public office for the purpose of obtaining or retaining business or securing an improper business advantage. The UK Bribery Act and other anti-corruption laws also prohibit commercial bribery not involving government officials, and requesting or accepting bribes; and anti-money laundering laws prohibit engaging in certain transactions involving criminally-derived property or the proceeds of criminal activity.We and third parties we do business with, as well as our representatives and agents, will have direct or indirect interactions with officials and employees of government agencies or state-owned or affiliated universities or other entities (for example, to obtain necessary permits, licenses, patent registrations and other regulatory approvals), which increases our risks under the FCPA and other anti-corruption laws. We also engage contractors, consultants and other third parties from time to time to conduct business development activities abroad. We may be held liable for the corrupt or other illegal activities of our employees or third parties even if we do not explicitly authorize such activities.
50
The FCPA also requires that we keep accurate books and records and maintain a system of adequate internal controls. We have recognized five material weaknesses in our internal control over financial reporting, which may compromise our ability to detect inappropriate payments (see the risk factor associated with “material weaknesses in our internal control over financial reporting”). Furthermore, we cannot assure you that our employees, agents, representatives, business partners or third-party intermediaries will always comply with our policies and applicable law, for which we may be ultimately held responsible.
Any allegations or violation of the FCPA or other applicable anti-bribery, anti-corruption laws and anti-money laundering laws may result in whistleblower complaints, sanctions, settlements, investigations, prosecution, enforcement actions, substantial criminal fines and civil penalties, disgorgement of profits, imprisonment, debarment, tax reassessments, breach of contract and fraud litigation, loss of export privileges, suspension or debarment from U.S. government contracts, adverse media coverage, reputational harm and other consequences, all of which may have an adverse effect on our reputation, business, financial condition, results of operations and prospects. Responding to an investigation or action can also result in a materially significant diversion of management’s attention and resources and significant defense costs and other professional fees.
Risks Related to Our Manufacturing Platform
We are designing an mRNA commercial manufacturing process in parallel with our product and process development. We currently intend to use contract development and manufacturing organizations (“CDMOs”), such as Samsung Biologics Co., Ltd., to produce material for
our COVID-19 product
candidate for late-stage clinical trials and commercial launch. There is risk that the final manufacturing process and facility could be incompatible with the CDMO facility, requiring modification and resulting in delays and inefficient deployment of capital.In an effort to bring our mRNA-related product candidates, particularlycandidate, to market more quickly, we are designing some aspects of our manufacturing process in parallel with selecting the exact manufacturing equipment and CDMO for that process. Steps to build the infrastructure include design, engineering, site selection and equipment procurement.
our pre-clinical COVID-19 vaccine
As we seek to increase the manufacturing output for commercial production and particular programs from the smaller quantities needed
for IND-enabling studies
to the larger quantities needed for commercial production, we intend to seek to continuously improve the manufacturability of our product candidates. Accordingly, we may change our manufacturing processes for a particular program during the course of development. However, any change in the manufacturing process may require resupplying clinical material to trial sites, which could increase our expenses, delay completion of clinical trials or otherwise adversely affect commercialization of the product.We plan to acquire additional laboratory, manufacturing and other space to accommodate our expected growth. If we are not able to access appropriate or sufficient space at reasonable cost, our business and results of operations could be adversely affected.
Our business and results of operations are dependent on locating and successfully negotiating leases for adequate access to laboratory and office space and suitable physical infrastructure, including electrical, plumbing, HVAC and network infrastructure, to conduct our operations and accommodate our growth. These resources are constrained and expensive in the areas in which we operate. If we are unable to access enough space or experience failures of our physical infrastructure, our business and results of operations could be adversely affected.
In order to properly conduct our business, we need access to sufficient laboratory space and equipment to perform the activities necessary to advance and complete our programs. Additionally, we need to ensure that our laboratories and corporate offices remain operational at all times, which includes maintaining suitable physical infrastructure, including electrical, plumbing and HVAC, logistics and transportation systems and network
51
infrastructure. We lease our laboratories and office spaces, and we rely on the landlords for basic maintenance of our leased laboratories and office buildings. If one of our landlords has not maintained a leased property sufficiently, we may be forced into an early exit from the facility, which could be disruptive to our business.
Our dsRNA and mRNA product candidates are based on innovative technologies and any product candidates we develop may be more complex and more difficult to manufacture than initially anticipated. We may encounter difficulties with manufacturing processes, manufacturing at higher volumes, product releases, product shelf life and storage, supply chain management, or shipping for any of our medicines, for both our agricultural or human health product candidates, including
our COVID-19 vaccine.
If we or any of our third-party vendors encounter such difficulties, our ability to supply commercial product or material for clinical trials could be delayed or stopped.The manufacturing processes for our product candidates using our dsRNA and mRNA platforms are innovative and complex. There are no mRNA medicines currently manufactured at commercial scale utilizing our manufacturing process. Due to the nature of this technology and our limited experience at commercial scale production, we could encounter difficulties with manufacturing processes, manufacturing at higher volumes, product releases, product shelf life and storage, supply chain management, or shipping.
These difficulties could be due to any number of reasons including, but not limited to, complexities of producing batches at any volume, equipment failure, choice and quality of raw materials and excipients, analytical testing technology, and product instability. In an effort to optimize product features, we may make changes to a product candidate in our manufacturing and stability formulation and conditions, which may result in our having to resupply batches
for pre-clinical or
clinical activities when there is insufficient product stability during storage and insufficient supply. Insufficient stability or shelf life of our product candidates could materially delay our or our strategic collaborators’ ability to continue the clinical trial for that product candidate or require us to begin a new clinical trial with a newly formulated drug product, due to the need to manufactureadditional pre-clinical or
clinical supply.Due to the nature of our products and manufacturing platform, there may also be a high degree of technological change that can negatively impact product comparability during and after clinical development. Furthermore, technology changes may drive the need for changes in, modification to, or the sourcing of new manufacturing infrastructure or may adversely affect third-party relationships.
The process to generate dsRNA or mRNA product candidates encapsulated in LNPs is complex and, if not developed and manufactured under well-controlled conditions, can adversely impact pharmacological activity and may result in one or more of our product candidates’ failure.
We have limited experience in manufacturing or commercializing proposed product candidates and may encounter difficulties, delays or other unanticipated hurdles before we are able to begin manufacturing our product candidates in the quantities needed to achieve our business plans.
We have limited experience in manufacturing or commercializing our proposed product candidates. As we increase manufacturing volume, we may encounter unexpected difficulties and delays that require adjustments or changes to our manufacturing process. Changes in our manufacturing processes may lead to failure of batches, which could lead to a substantial delay
in pre-clinical studies
and clinical trials or the delivery of commercial product. Any such changes could adversely affect clinical or commercial supply of our products. Such changes might also cause delays in or increase the cost of commissioning our facility and adversely affect our commercialization plans.We have increased the batch size for our mRNA production to accommodate the supply requirements of some of our current and
anticipated pre-clinical and
clinical programs. However, in some cases, we may have to52
utilize multiple batches of substance and product to meet the clinical supply requirement of a single clinical trial. If we or our contract manufacturers fail to successfully and consistently produce mRNA at larger batch sizes, it could lead to a substantial delay in our clinical trials or in the commercialization of any products that may receive regulatory approval.
If our cell-free manufacturing platform does not perform as expected, or if our competitors develop and market technologies or products more rapidly than we do or that are more effective, outperform, safer or less expensive than our manufacturing platform technology, our commercial opportunities will be negatively impacted.
It is anticipated that we will face increased competition in the future as new companies enter the markets and as scientific developments progress. If we are unable to compete effectively, our opportunity to discover new products or generate revenue from the sale of such new products or our existing product candidates could be adversely affected.
We have established laboratory, clean room, and manufacturing facilities in Massachusetts, North Carolina and New York to support our activities, which is resulting in the incurrence of significant investment with no assurance that such investment will be recouped.
In order to support our future growth and our agriculture and human health product pipeline, we have invested in facilities to develop products or produce materials. This investment has significantly increased our capital and operating expenses. Moreover, based on our current business plan, we anticipate that in the future we will need to expand our facilities for research and development and production capacity, which we currently expect to accomplish by expanding the capacity of existing facilities. We may need to, or decide to, build additional commercial mRNA manufacturing facilities using our platform technology in the U.S. and in countries outside of the U.S. There can be no assurance that any additional manufacturing capacity that we may acquire will be necessary or that this investment will be recouped.
If we are unable to adequately and timely manufacture and supply our products and product candidates or if we do not fully utilize our manufacturing facilities, our business may be adversely affected. Charges resulting from excess capacity would have a negative effect on our financial condition and results of operations.
We will depend on relationships with third parties for revenues, and for the development, regulatory approval, commercialization and marketing of certain of our products and product candidates, which are outside of our full control.
We rely on a number of third-party relationships for the development, regulatory approval and commercialization of certain of our product candidates, including, but not limited to, the Azzur Group for our cleanroom. Certain aspects of our regulatory affairs and clinical development relating to our products and product candidates are also outsourced to third parties. Reliance on third parties subjects us to a number of risks, including:
| • | the inability to control the resources such third parties devote to our programs, products or product candidates; |
| • | disputes may arise under an agreement and the underlying agreement may fail to provide us with significant protection or may fail to be effectively enforced if such third parties fail to perform; |
| • | failure or perceived failure by us to comply with our obligations under an agreement with a third party could cause the third party to make public statements against us or could result in litigation asserting that we have breached our obligations, any of which could have a material adverse effect on our reputation, business, financial condition, or results of operations; |
| • | the interests of such third parties may not always be aligned with our interests, and such parties may not pursue regulatory approvals or market a product in the same manner or to the same extent that we |
53
would, which could adversely affect revenues, or may adopt tax strategies that could have an adverse effect on our business, results of operations or financial condition; |
| • | third-party relationships require the parties to cooperate, and failure to do so effectively could adversely affect product development or the clinical development or regulatory approvals of product candidates under collaborative control, could result in termination of the research, development or commercialization of product candidates or could result in litigation or arbitration; |
| • | any failure on the part of such third parties to comply with applicable laws, including tax laws, regulatory requirements and/or applicable contractual obligations or to fulfill any responsibilities they may have to protect and enforce any intellectual property rights underlying product candidates could have an adverse effect on revenues as well as give rise to possible legal proceedings; and |
| • | any improper conduct or actions on the part of such third parties could subject us to civil or criminal investigations and monetary and injunctive penalties, impact the accuracy and timing of financial reporting and/or adversely impact our ability to conduct business, operating results and reputation. |
Given these risks, there is considerable uncertainty regarding the success of current and future collaborative efforts. If these efforts fail, our product development or commercialization of product candidates could be delayed, revenues from products could decline and/or the anticipated benefits of these arrangements may not be realized.
Manufacturing issues could substantially increase costs, limit supply of products and/or reduce revenues.
The process of manufacturing our products is complex, highly regulated and subject to numerous risks, including:
| • | Risks of Reliance on Third Parties and Single Source Providers. the COVID-19 pandemic and intellectual property protection. These third parties may not perform their obligations in a timely and cost-effective manner or in compliance with applicable regulations, and they may be unable or unwilling to increase production capacity commensurate with demand for existing or future products. Finding alternative providers could take a significant amount of time and involve significant expense due to the specialized nature of the services and the need to obtain regulatory approval of any significant changes to suppliers or manufacturing methods. We cannot be certain that we could reach an agreement with alternative providers or that the FDA or other regulatory authorities would approve the use of such alternatives. |
| • | Risks Relating to Compliance with cGMP. |
| • | Global Supply Risks. |
54
adversely affected by equipment failures, labor shortages, public health epidemics, natural disasters, power failures, cyber-attacks and many other factors. Additionally, there can be no assurance that we will be able to meet expected timelines or that there will not be any direct or indirect delays resulting from the COVID-19 pandemic. We have had delays, and might experience additional delays, in bringing our current and planned facilities online, and we may not have sufficient manufacturing capacity to meet our long-term manufacturing requirements. |
| • | Risk of Product Loss. |
Any adverse developments affecting manufacturing operations or the operations of third-party suppliers and manufacturers may result in shipment delays, inventory shortages, lot failures, product withdrawals or recalls or other interruptions in the commercial supply of our products. Additionally, we may also have to take inventory write-offs and incur other charges and expenses for products that fail to meet specifications, undertake costly remediation efforts or seek more costly manufacturing alternatives. Such developments could increase manufacturing costs, cause revenue loss or loss of market share as patients and physicians turn to competing therapeutics, diminish profitability or damage our reputation.
In addition, although we have business continuity plans to reduce the potential for manufacturing disruptions or delays and reduce the severity of a disruptive event, there is no guarantee that these plans will be adequate, which could adversely affect our business and operations.
Risks Related to the Production of dsRNA and mRNA
Our proposed products are temperature sensitive and may have other attributes that lead to limited shelf life. These attributes may pose risks to supply, inventory and waste management and increased cost of goods.
Our mRNA and dsRNA product candidates may prove to have a stability profile that leads to a lower than desired shelf life of the final approved mRNA medicine. This poses risk in supply requirements, wasted stock, and higher cost of goods.
Our products and product candidates are temperature sensitive, and we may learn that any or all of our product candidates are less stable than desired. It is also possible that we may find that transportation conditions negatively impact product quality. This may require changes to the formulation or manufacturing process for one or more of our product candidates and result in delays or interruptions to clinical or commercial supply. In addition, the cost associated with such transportation services and the limited pool of vendors may also add additional risks of supply disruptions.
We have established a number of analytical assays, and may have to establish several more, to assess the quality of our dsRNA and mRNA product candidates. We may identify gaps in our analytical testing strategy that might prevent release of product or could require product withdrawal or recall. For example, new impurities that have an impact on product safety, efficacy, or stability may be discovered. This may lead to an inability to release mRNA product candidates until the manufacturing or testing process is rectified.
55
Risks Related to Raw Materials and Reliance on Third Parties
The materials used in the processes by which we manufacture RNA and our derivative products, such as dsRNA or mRNA, may become difficult to obtain in the quality or quantity required for our business plans or at the prices that are currently projected.
Many of our processes and products rely on materials purchased from third parties and should these materials increase in prices, have supply constraints, or become unavailable, it could impact our ability to develop products or bring them to market either on time, at competitive prices or at all. For example, our dsRNA processes use specific yeast microbial RNA, the most effective forms of which are sourced from suppliers in China. Should that particular yeast become unavailable, it could impair the effectiveness, yield or availability of the dsRNA produced for the agricultural markets.
Moreover, some enzymes that are used in our RNA platform and our down-stream products are specific in nature and sourced from third parties, some of whom have proprietary processes which give them an advantage in cost or effectiveness that they pass on to us. Some materials come from single sources, such as LNPs, which are licensed from a third party and which are used to produce mRNA. We may need to license other materials from third parties, and if we are unable to do so on reasonable terms, or at all, it could have a material adverse effect on our business.
Some of the raw materials are being employed in an innovative manner and may not be scaled to a level to support commercial supply and we could experience unexpected manufacturing or testing failures, or supply shortages. Such issues with raw materials and excipients could cause delays or interruptions to clinical and commercial supply of products or product candidates.
Single or limited sources for some materials may impact our ability to secure supply.
Our dependence on single-source, limited-source or preferred suppliers exposes us to certain risks, such as:
| • | a disruption to suppliers’ operations which could leave us with no other means of continuing the research, development, or manufacturing operations for which the supplier provides inputs; |
| • | the inability to locate a suitable replacement on acceptable terms or on a timely basis, if at all; |
| • | existing suppliers may cease or reduce production or deliveries, raise prices, or renegotiate terms; |
| • | delays caused by supply issues may harm our reputation, frustrate customers, and cause them to turn to our competitors; and |
| • | Our ability to progress the development of existing programs and the expansion of capacity to begin future programs could be materially and adversely impacted if the single-source, limited-source or preferred suppliers upon which we rely were to experience a significant business challenge, disruption, or failure due to issues such as financial difficulties or bankruptcy, issues relating to other customers such as regulatory or quality compliance issues, or other financial, legal, regulatory, or reputational issues. |
Should any of the above risks, or should any consequences of unpredictable risks, come to fruition, such events could have a material adverse effect on operations.
We rely on highly specialized equipment and consumables for the production of RNA and our derivative products, dsRNA and mRNA, and any disruption to the supply chain or any malfunction of that equipment may adversely impact our operations.
The equipment and consumables used to produce RNA and our derivative products, dsRNA and mRNA, are currently supply constrained across all suppliers, which may cause delays in development, testing or marketing of our human health products and may require us to ultimately increase prices should our products become available to consumers.
56
Additionally, we will be dependent on a number of equipment providers and CMOs who are also implementing innovative technology. If such equipment malfunctions or if we encounter unexpected performance issues, we could encounter delays or interruptions to clinical and commercial supply. Due to the number of different programs, we may have cross contamination of product candidates inside of our factories, CROs, suppliers, or in the clinic that affect the integrity of our product candidates.
Delay or unavailability of products, services, or equipment provided by suppliers could require us to change the design of our research, development, and manufacturing processes based on the functions, limitations, features, and specifications of the replacement items or seek out a new supplier to provide these items. Additionally, as we grow, our existing suppliers may not be able to meet our increasing demand, and additional suppliers may need to be found. We may not be able to secure suppliers who provide lab supplies at, or equipment and services to, the specification, quantity, and quality levels that we demand (or at all) or be able to negotiate acceptable fees and terms of services with such suppliers.
Risks Related to Market Acceptance
Even if any of the product candidates we develop receives regulatory approval, we may nonetheless fail to achieve the degree of market acceptance by physicians, patients, healthcare payors, and others in the medical community necessary for commercial success.
The commercial success of any of our product candidates will depend upon its degree of market acceptance by physicians, patients, third party payors, and others in the medical community. Even if any product candidates we develop receives regulatory approval, they may nonetheless fail to gain sufficient market acceptance by physicians, patients, healthcare payors, and others in the medical community. The degree of market acceptance of any product candidates we may develop, if approved for commercial sale, will depend on a number of factors, including:
| • | the wider acceptance by patients of products derived from RNA manufacturing processes; |
| • | the efficacy and safety of such product candidates as demonstrated in pivotal clinical trials published in peer-reviewed journals; |
| • | the potential and perceived advantages compared to alternative treatments; |
| • | the ability to offer our products for sale at competitive prices; |
| • | the ability to offer appropriate patient access programs, such as co-pay assistance; |
| • | the extent to which physicians recommend our products to their patients; |
| • | convenience and ease of dosing and administration compared to alternative treatments; |
| • | the clinical indications for which the product candidate is approved by the FDA, EMA or other comparable foreign regulatory agencies; |
| • | product labeling or product insert requirements of the FDA, EMA or other comparable foreign regulatory authorities, including any limitations, contraindications or warnings contained in a product’s approved labeling; |
| • | restrictions on how the product is distributed; |
| • | the timing of market introduction of competitive products; |
| • | publicity concerning our products or competing products and treatments; |
| • | the effectiveness of marketing and distribution efforts by us and other licenses and distributors; |
| • | sufficient governmental third party coverage or reimbursement; and |
| • | the prevalence and severity of any side effects. |
57
If any product candidates developed by us does not achieve an adequate level of acceptance by physicians, healthcare payors, patients and the medical community, we will not be able to generate significant revenue, and may not become or remain profitable. The failure of any product candidates to find market acceptance could harm our business prospects.
Legal requirements as well as ethical and social concerns about synthetic biology and genetic engineering could limit or prevent the use of our technologies and limit revenues.
Our platform technology, including how dsRNA and mRNA is extracted, includes the use of synthetic biology and genetic engineering. In some countries, drugs made using genetically modified organisms may be subject to a more stringent legal regime, which could prove to be complex and very challenging. For example, in the European Union, the rules on genetically modified organisms could apply in addition to the general rules on medicinal products or cosmetic products. The rules on advanced therapy medicinal products may also apply.
Additionally, public perception about the safety and environmental hazards of, and ethical concerns over, synthetic biology and genetic engineering could influence public acceptance of our technologies, product candidates and processes, particularly in the case of human medicines such as
our COVID-19 vaccine
product candidate. If we, our collaborators or other third parties are not able to overcome the legal challenges as well as the social concerns relating to synthetic biology and genetic engineering, our technologies, product candidates and processes may not be accepted. These challenges and concerns could result in increased expenses, regulatory scrutiny and increased regulation, trade restrictions on imports of our product candidates, delays or other impediments to our programs or the public acceptance and commercialization of our products. We design and produce product candidates with characteristics comparable or superior to those found in naturally occurring organisms or enzymes in a controlled laboratory; however, the release of such organisms into uncontrolled environments could have unintended consequences. Any adverse effect resulting from such a release could have a material adverse effect on our business, financial condition or results of operations, and may expose us to liability for any resulting harm.Risks Related to Global Expansion
Our planned manufacturing, sales and operations are subject to the risks of doing business internationally.
In the future, we intend to expand the reach of our platform technology into international markets, including certain countries in Africa, Asia and Latin America where the need for locally produced vaccines is high, subjecting us to many risks that could adversely affect our business and revenues. There is no guarantee that our efforts and strategies to expand manufacturing and sales in international markets will succeed. Emerging market countries may be especially vulnerable to periods of global and local political, legal, regulatory and financial instability and may have a higher incidence of corruption and fraudulent business practices. Certain countries may require local clinical trial data as part of the drug registration process in addition to global clinical trials, which can add to overall drug development and registration timelines. We may also be required to increase our reliance on third-party agents and unfamiliar operations and arrangements previously utilized by companies we collaborate with or acquire in emerging markets.
Our manufacturing, sales and operations are subject to the risks of doing business internationally, including:
| • | difficulties and challenges relating to the building, commissioning and complying with regulatory requirements related to manufacturing facilities in foreign countries; |
| • | the inability to obtain necessary foreign regulatory or pricing approvals of products in a timely manner; |
| • | limitations and additional pressures on our ability to obtain and maintain product pricing or receive price increases, including those resulting from governmental or regulatory requirements; |
| • | the inability to successfully complete preclinical studies or subsequent or confirmatory clinical trials in countries where our experience is limited; |
58
| • | the willingness of regulatory agencies to accept data from preclinical studies or clinical trials conducted in other jurisdictions; |
| • | longer payment and reimbursement cycles and uncertainties regarding the collectability of accounts receivable; |
| • | fluctuations in foreign currency exchange rates that may adversely impact our revenues, net income and value of certain of our investments; |
| • | the imposition of governmental controls; |
| • | diverse data privacy and protection requirements; |
| • | increasingly complex standards for complying with foreign laws and regulations that may differ substantially from country to country and may conflict with corresponding U.S. laws and regulations; |
| • | the far-reaching anti-bribery and anti-corruption legislation in the U.K., including the Bribery Act, and elsewhere and escalation of investigations and prosecutions pursuant to such laws; |
| • | compliance with complex import and export control laws; |
| • | changes in tax laws; |
| • | the imposition of tariffs or embargoes and other trade restrictions; |
| • | the impact of public health epidemics, such as the COVID-19 pandemic, on the global economy and the delivery of healthcare treatments; |
| • | less favorable intellectual property or other applicable laws; and |
| • | known and unknown risks related to local and geopolitical unrest; |
In addition, our future potential international operations are subject to regulation under U.S. law,
and non-compliance with
those laws may subject us to severe criminal and civil penalties. For example, the FCPA prohibits U.S. companies and their representatives from paying, offering to pay, promising to pay or authorizing the payment of anything of value to any foreign government official, government staff member, political party or political candidate for the purpose of obtaining or retaining business or to otherwise obtain favorable treatment or influence a person working in an official capacity. In many countries, the health care professionals with whom we may regularly interact may meet the FCPA’s definition of a foreign government official. Failure to comply with domestic or foreign laws could result in various adverse consequences, including possible delay in approval or refusal to approve a product, recalls, seizures or withdrawal of an approved product from the market, disruption in the supply or availability of our products or suspension of export or import privileges, the imposition of civil or criminal sanctions, the prosecution of executives overseeing our international operations and damage to our reputation. Any significant impairment of our ability to sell products outside of the U.S. could adversely impact our business and financial results.Our goal of expanding outside the U.S. will depend on our ability to successfully manage the complexity of multiple global supply chains in countries with poor infrastructure, in which we have limited experience.
Logistics, regulatory environments, business customs, local and geopolitical concerns and end user markets differ country by country. As we expand globally to enable the production of our dsRNA and mRNA products in countries outside the U.S., we will face material risks that could cause us to expend significant resources. There can be no guarantee when such efforts will be successful, if at all. As we expand our platform globally, we will have to familiarize ourselves with the regulatory environment in that country, which could significantly diverge from the regulatory regimes in the U.S. and which may not necessarily approve our product candidates, even if such product candidates achieve regulatory approval in the U.S.
In each country in which we intend to utilize our manufacturing platform, it may also be necessary to create partnerships with local enterprises, which come with inherent risks, including corruption, violation of U.S. laws
59
and regulations relating to anti-corruption laws, intellectual property theft, divergence from our quality and health standards and a number of unknown risks that could delay or cause our international expansion to fail entirely.
We will have to become familiar with these and other factors in order to be effective; however, our ability to do so is untested as is our ability to obtain and retain experts in these areas to implement an international supply chain serving any facility other than those in the United States. Moreover, any of our suppliers could go out of business, lose operating licenses, be subject to regulatory actions and be unable to supply us, which could result in production delays or stoppages.
Risks Related to Competition
We face significant competition, and if our competitors develop and market technologies or products more rapidly than we do or that are more effective, safer or less expensive than the product candidates we develop, our commercial opportunities will be negatively impacted.
We believe one of our key competitive advantages is the cost and quality at which we can make RNA and recover dsRNA from it for use in agricultural products or mRNA for use in human health products. Should other processes match or beat that cost or quality, we could lose a key competitive advantage as an RNA producer which could in turn have negative effects on the products in our pipeline which depend on the quality and cost of the RNA produced by us to be competitive. Fermentation is currently the most popular method competing with our process for the production of RNA. While we believe fermentation is currently more expensive and tends to produce more down-stream impurities than our proprietary process, innovation or scale in the fermentation process could cause these drawbacks to be overcome to produce a product that is cost competitive with ours. Conventional cell-free processes, such
as in-vitro transcription
are cost prohibitive for agricultural applications and require special inputs. New innovations in cell-free processes could outperform our cell-free processes and make our technology obsolete.Rapidly changing technology and emerging competition in the synthetic biology industry could make the platform, programs, and products we are developing obsolete
or non-competitive unless
development of our platform and pursuit of new market opportunities continues.The synthetic biology industry is still emerging and is characterized by rapid and significant technological changes, frequent new product introductions and enhancements, and evolving industry demands and standards. Our future success will depend on our ability to develop, manufacture and commercialize our product candidates on a timely and cost-effective basis.
Our competitors, either alone or together with collaborators, may have significantly greater financial, manufacturing, marketing, drug development, technical and human resources and commercial expertise than we do. Our competitors may also have more experience:
| • | developing drug candidates; |
| • | conducting preclinical and clinical trials; |
| • | obtaining regulatory approvals; and |
| • | commercializing product candidates. |
60
Risks Related to our Human Health Program
Even if we successfully design and complete our preclinical studies, our preclinical human health product candidates, and similar products in the future, must still go through clinical studies, which may reveal significant adverse events, including negative immune system responses, and may result in a safety profile that could prevent or delay regulatory approval or licensure or market acceptance of candidate products.
There is typically an extremely high rate of attrition for product candidates across categories of medicines proceeding through clinical trials. These product candidates may fail to show the desired safety and efficacy profile in later stages of clinical trials despite having progressed through nonclinical studies and initial clinical trials. A number of companies in the biopharmaceutical industry have suffered significant setbacks in later stage clinical trials due to lack of efficacy or unacceptable safety profiles, notwithstanding promising results in earlier trials. Most investigational medicines that commence clinical trials are never approved as products and there can be no assurance that any of our current or future clinical trials will ultimately be successful or support further clinical development of any of our investigational medicines. Certain aspects of our investigational medicines may induce immune reactions from either the mRNA or the lipid as well as adverse reactions within liver pathways or degradation of the mRNA or the lipid nanoparticle LNP, any of which could lead to significant adverse events in one or more of our clinical trials. Many of these types of side effects have been seen for previously developed LNPs. There may be resulting uncertainty as to the underlying cause of any such adverse event, which would make it difficult to accurately predict side effects in future clinical trials and would result in significant delays in our programs.
If unacceptable side effects, including materialized risks of immunogenicity, arise in the development of our product candidates, the FDA, a comparable foreign regulatory authority or the Institutional Review Boards (IRBs) at the institutions in which its studies are conducted, or the Data Safety Monitoring Board, if constituted for its clinical studies could recommend a suspension or termination of our clinical studies, or the FDA or comparable foreign regulatory authorities could order us to cease further development of or deny licensure or approval of a product candidate. In addition, side effects could affect patient recruitment or the ability of enrolled patients to complete a trial or result in potential product liability claims. In addition, these side effects may not be appropriately recognized or managed by the treating medical staff. We expect to have to train medical personnel using our product candidates to understand the side effect profiles for our clinical studies and upon any commercialization of any of our product candidates. Inadequate training in recognizing or managing the potential side effects of our product candidates could result in patient injury or death.
Even if such side effects do not preclude the drug from obtaining or maintaining marketing approval, any such approval may be for a more narrow indication than we seek or an unfavorable benefit risk ratio may inhibit market acceptance of the approved product due to its tolerability versus other therapies. In addition, regulatory authorities may not approve the labeling claims that are necessary or desirable for the successful commercialization of any product candidates we develop. Consequently, the commercial prospects of such product candidates may be harmed, and our ability to generate product revenues from any of these product candidates may be delayed or eliminated. Any of these occurrences may harm our business, financial condition and prospects significantly.
Additionally, if one or more of our product candidates receives marketing licensure and/or approval, and we or others later identify undesirable side effects caused by such products, a number of potentially significant negative consequences could result, including:
| • | regulatory authorities may withdraw licensures and/or approvals of such product; |
| • | regulatory authorities may require additional warnings on the label, such as a “black box” warning or contraindication; |
| • | additional restrictions may be imposed on the marketing of the particular product or the manufacturing processes for the product or any product component; |
61
| • | we may be required to restrict the conductions under which the product may be distributed, including through implementation of a Risk Evaluation and Mitigation Strategy, or REMS; |
| • | we may be required to change the way a product candidate is administered or conduct additional clinical trials; |
| • | we could be sued and held liable for harm caused to patients; |
| • | the product may become less competitive; and |
| • | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of a product candidate, if approved, and could significantly harm our business, results of operations and prospects.
Our human health markets are highly competitive. If we are unable to compete effectively with existing products, new treatment methods and new technologies, we may be unable to commercialize any products that we may develop in the future.
The biotechnology market is highly competitive, is subject to rapid technological change and is significantly affected by existing rival products and medical procedures, new product introductions and the market activities of other participants. Pharmaceutical and biotechnology companies, academic institutions, governmental agencies and other public and private research organizations may pursue the research and development of technologies, products or other therapeutic products for the treatment of some or all of the diseases that we target. We also may face competition from products that have already been approved or licensed and accepted by the medical community for the treatment of these same indications. Our competitors may develop products more rapidly or more effectively than us. Many of our competitors have:
| • | much greater experience, financial, technical and human resources than we have at every stage of the discovery, development, manufacture and commercialization process; |
| • | more extensive experience in preclinical studies, conducting clinical trials, obtaining and maintaining regulatory approvals or licensures and manufacturing and marketing products; |
| • | products that have been approved or licensed or are in late stages of development; |
| • | established distribution networks; |
| • | collaborative arrangements with leading companies and research institutions; and |
| • | entrenched and established relationships with healthcare providers and payors. |
In addition, many of these companies, in contrast to us, are well-capitalized. As a result of any of the foregoing factors, our competitors may develop or commercialize products with significant advantages over any product that we may develop in the future. If our competitors are more successful in commercializing their products than us, their success could adversely affect our competitive position and harm our business prospects.
If we encounter difficulties enrolling patients in our clinical studies, including due
to COVID-19, our
clinical development activities could be delayed or otherwise adversely affected.We may experience difficulties in patient enrollment in our clinical studies for a variety of reasons. The timely completion of clinical studies in accordance with their protocols depends, among other things, on our ability to enroll a sufficient number of patients who remain in the trial until its conclusion. The enrollment of patients depends on many factors, including:
| • | the patient eligibility and exclusion criteria defined in the protocol; |
| • | the severity of the disease under investigation; |
62
| • | the size of the patient population required for analysis of the trial’s primary endpoints and the process for identifying patients; |
| • | the proximity of patients to trial sites; |
| • | the design of the trial; |
| • | our ability to recruit clinical study investigators with the appropriate competencies and experience; |
| • | clinicians’ and patients’ perceptions as to the potential advantages and risks of the product candidate being studied with respect to other available therapies, including any new products that may be approved for the indications we are investigating; |
| • | the availability of competing commercially available therapies and other competing product candidates’ clinical studies; |
| • | the ability to monitor patients adequately during and after treatment; |
| • | efforts to facilitate timely enrollment in clinical trials; |
| • | our ability to obtain and maintain patient informed consents; and |
| • | the risk that patients enrolled in clinical studies will drop out of the trials before completion. |
Further, timely enrollment in clinical studies is reliant on clinical study sites, which may experience delays or otherwise be adversely affected by disruptions due to global health matters,
including COVID-19 and
other pandemics.If successfully released for sale,
our COVID-19 vaccine
candidate may fail to gain market acceptance.Even if our mRNA vaccine
for COVID-19 successfully
completes clinical trials and is approved for commercial marketing, it may fail to meet the same high level of efficacy demonstrated by competitors and our ability to obtain revenues from the vaccine may be diminished or eliminated altogether. Moreover, the addressable market forour COVID-19 candidate
may be limited if competing products have saturated some or all markets, particularly the most profitable markets.We may incur additional costs or experience delays in completing the development and commercialization of our product candidates.
Clinical testing is expensive, difficult to design and implement, can take many years to complete and is uncertain as to timing and outcome. A failure of one or more clinical studies can occur at any stage in the process. We may experience delays in initiating or completing clinical studies. We may also experience numerous unforeseen events during, or as a result of, any future clinical studies that could delay or prevent our ability to receive marketing licensure and approval to commercialize our product candidates, including:
| • | Regulators, or IRBs, Data Safety Monitoring Boards, or ethics committees may not authorize us or our investigators to commence a clinical study or conduct a clinical study at a prospective trial site or may impose burdensome restrictions on or cause delays of the clinical study at a prospective trial site; |
| • | the FDA or other comparable regulatory authorities may disagree with our clinical study design, including with respect to dosing levels administered in our planned clinical studies, which may delay or prevent us from initiating our clinical studies with our originally intended trial design; |
| • | the FDA or other comparable regulatory authorities may not accept the results of any of our clinical studies conducted in other geographic locations or outside their country’s jurisdiction; |
| • | we may experience delays in reaching, or fail to reach, agreement on acceptable terms with prospective trial sites and prospective Contract Research Organizations (CROs), which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
63
| • | the number of subjects required for clinical studies of any product candidates may be larger than we anticipate or subjects may drop out of these clinical studies or fail to return for post-treatment follow-up at a higher rate than we anticipate; |
| • | our third-party contractors may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all, or may deviate from the clinical study protocol or drop out of the trial, which may require that we add new clinical study sites or investigators; |
| • | we may experience delays and interruptions to clinical studies, we may experience delays or interruptions to our manufacturing supply chain, or we could suffer delays in reaching, or we may fail to reach, agreement on acceptable terms with third-party service providers on whom we rely; |
| • | additional delays and interruptions to our clinical studies could extend the duration of the trials and increase the overall costs to finish the trials as its fixed costs are not substantially reduced during delays; |
| • | we may elect to, or regulators, IRBs, Data Safety Monitoring Boards or ethics committees may require that we or our investigators, suspend or terminate clinical research or trials for various reasons, including noncompliance with regulatory requirements or a finding that the participants are being exposed to unacceptable health risks; |
| • | we may need to amend or submit new clinical protocols because of changes in regulatory requirements and guidance; |
| • | we may not have the financial resources available to begin and complete the planned trials, or the cost of clinical studies of any product candidates may be greater than we anticipate; and |
| • | the supply or quality of our product candidates or other materials necessary to conduct clinical studies of our product candidates may be insufficient or inadequate to initiate or complete a given clinical study. |
Our product development costs will increase if we experience additional delays in clinical testing or in obtaining marketing approvals or licensure. We do not know whether any of our clinical studies will begin as planned, will need to be restructured or will be completed on schedule, or at all. If we do not achieve our product development goals in the time-frames we announce and expect, the approval, licensure, and commercialization of our product candidates may be delayed or prevented entirely. Significant clinical study delays also could shorten any periods during which we may have the exclusive right to commercialize our product candidates and may allow our competitors to bring products to market before we do, potentially impairing our ability to successfully commercialize our product candidates and harming our business and results of operations. Any delays in our clinical development programs may harm our business, financial condition and results of operations significantly.
The time and expense required to obtain regulatory approvals for our preclinical and clinical trials could be significantly greater than for more conventional therapeutic technologies or products. If clinical trials of any product candidates fail to demonstrate safety and efficacy to the satisfaction of regulatory authorities or do not otherwise produce positive results, we may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of such product candidates.
In the United States, the products that we intend to develop and market are regulated by the FDA under its drug and biologic development and review processes. Before such products can be marketed, we must obtain multiple authorizations and licensures from the FDA, first through submission and authorization of an investigational new drug application, or IND, then through successful completion of human testing under three phases of clinical trials and finally through submission of a biologics license application, or BLA. Even after successful completion of clinical testing, there is a risk that the FDA may refuse to file our BLA submissions, request further information from us, disagree with our findings, undertake a lengthy review of our BLA submissions or deny our application.
64
The time required to obtain approval or licensure by the FDA and other comparable regulatory authorities is unpredictable but typically takes many years following the commencement of clinical trials, if at all, and depends upon numerous factors, including the substantial discretion of the regulatory authorities. In addition, approval and licensure policies, regulations or the type and amount of clinical data necessary to gain approval or licensure may change during the course of a product candidate’s clinical development and may vary among jurisdictions. We have not obtained marketing approval or licensure for any product candidate and it is possible that none of our existing product candidates, or any product candidates we may seek to develop in the future, will ever obtain marketing approval or licensure.
Our product candidates could fail to receive BLA licensure in the United States for many reasons, including the following:
| • | the FDA may disagree with the design or implementation of our clinical trials; |
| • | we may be unable to demonstrate to the satisfaction of the FDA that a product candidate is safe, pure, and potent; |
| • | results of clinical trials may not meet the level of statistical significance required by the FDA for licensure; |
| • | we may be unable to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks; |
| • | the FDA may disagree with our interpretation of data from preclinical studies or clinical trials; |
| • | data collected from clinical trials of our product candidates may not be sufficient to support the submission of a BLA to the FDA or other submission or to obtain marketing licensure in the United States; |
| • | the FDA may find deficiencies with or fail to approve the manufacturing processes or facilities of third-party manufacturers with which we contract for clinical and commercial supplies; and |
| • | the licensure policies or regulations of the FDA may significantly change in a manner rendering our clinical data insufficient for licensure. |
This lengthy licensure process as well as the unpredictability of future clinical trial results may result in our failing to obtain BLA licensure to market any of our product candidates, which would significantly harm our business, results of operations, financial condition and prospects. The FDA has substantial discretion in the licensure process and determining when or whether regulatory licensure will be obtained for any of our product candidates. Even if we believe the data collected from clinical trials of our product candidates are promising, such data may not be sufficient to support licensure by the FDA.
In addition, even if we were to obtain licensure, regulatory authorities may approve any of our product candidates for fewer or more limited indications than we request, may not approve the price we intend to charge for our products, may grant a license contingent on the performance of costly post marketing clinical trials, or may approve or license a product candidate with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that product candidate. Any of the foregoing scenarios could materially harm the commercial prospects for our product candidates.
If we decide to market any product that we develop in jurisdictions in addition to the United States, we may incur the same costs or more in satisfying foreign regulatory requirements governing the conduct of preclinical and clinical trials, manufacturing and marketing and commercialization of any product that we develop in the future. Approval or licensure by the FDA by itself does not assure approval by regulatory authorities outside the United States. Each of these foreign regulatory approval processes includes all of the risks associated with the FDA approval or licensing process, as well as risks attributable to having to satisfy local regulations within each of these foreign jurisdictions. Our inability to obtain regulatory approval outside the United States may also adversely compromise our business prospects.
65
We may have difficulties convincing the medical community and third-party payors to accept and use any product that we are able to develop in the future even following our receipt of regulatory approval or licensure for commercialization. Key participants in pharmaceutical marketplaces, such as physicians, third-party payors and consumers, may not accept a product that we develop. Even if such a product is accepted by these participants, the medical community may not consider effectiveness and safety alone as a sufficient basis for prescribing such as product in lieu of other alternative treatment methods and medications that are available.
We may not be successful in our efforts to identify, develop, or commercialize potential product candidates.
The success of our business depends primarily upon our ability to identify, develop, and commercialize products based on our mRNA platform. Research programs to identify new product candidates require substantial technical, financial, and human resources. We may focus our efforts and resources on potential programs or product candidates that ultimately prove to be unsuccessful. As our development candidates and investigational medicines progress, we or others may determine that: certain of our risk allocation decisions were incorrect or insufficient; we made platform level technology mistakes; individual programs or our mRNA science in general have technology or biology risks that were unknown or under-appreciated; our choices on how to develop our infrastructure to support our needs would likely result in an inability to manufacture our investigational medicines for clinical trials or would otherwise impair our manufacturing; or we have allocated resources in such a way that large investments would not be recovered and capital allocation would not be subject to
rapid re-direction. All
of these risks may relate to our current and future programs sharing similar science (including mRNA science) and infrastructure, and in the event material decisions in any of these areas turn out to have been incorrect or under-optimized, we may experience a material adverse impact on our business and ability to fund our operations and we may never realize what we believe is the potential of mRNA. If any of these events occur, we may be forced to abandon our development efforts for one or more programs, which would have a material adverse effect on our business, financial condition, results of operations, and prospects.The successful commercialization of our human health product candidates will depend in part on the extent to which third-party insurers and payors establish adequate coverage, reimbursement levels and pricing policies. Failure to obtain or maintain coverage and adequate reimbursement for our product candidates, if approved, could limit our ability to market those products and decrease our ability to generate revenue.
The availability and extent of coverage and adequate reimbursement by third-party payors, including government health administration authorities, private health coverage insurers, managed care organizations and other third-party payors is essential for most patients to be able to afford expensive treatments. Sales of any of our product candidates that receive marketing approval will depend substantially, both in the United States and internationally, on the extent to which the costs of such product candidates will be covered and reimbursed by third-party payors. If reimbursement is not available, or is available only to limited levels, we may not be able to successfully commercialize our product candidates. Even if coverage is provided, the approved reimbursement amount may not be high enough to allow us to establish or maintain pricing sufficient to realize an adequate return on our investment. Coverage and reimbursement may impact the demand for, or the price of, any product candidate for which we obtain marketing approval. If coverage and reimbursement are not available or reimbursement is available only to limited levels, we may not successfully commercialize any product candidate for which we obtain marketing approval.
There is significant uncertainty related to third-party payor coverage and reimbursement of newly approved products. In the United States, for example, principal decisions about reimbursement for new products are typically made by the Centers for Medicare & Medicaid Services, or CMS, an agency within the U.S. Department of Health and Human Services. CMS decides whether and to what extent a new product will be covered and reimbursed under Medicare, and private third-party payors often follow CMS’s decisions regarding coverage and reimbursement to a substantial degree. However, one third-party payor’s determination to provide coverage for a product candidate does not assure that other payors will also provide coverage for the product candidate. Further, no uniform policy for coverage and reimbursement exists in the United States, and coverage
66
and reimbursement can differ significantly from payor to payor. As a result, the coverage determination process is often time-consuming and costly. This process will require us to provide scientific and clinical support for the use of our products to each third-party payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance.
We cannot predict the likelihood, nature, or extent of health reform initiatives that may arise from future legislation or administrative action.
Changes in healthcare policy could increase our costs, decrease our revenue and impact sales of and reimbursement for our future products, if approved. We expect to experience pricing pressures in connection with the sale of our product candidates due to the trend toward managed health care, the increasing influence of health maintenance organizations and additional legislative changes. The United States and some foreign jurisdictions are considering or have enacted a number of legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to sell our products profitably once approved. Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality or expanding access. Current and future legislative proposals to further reform healthcare or reduce healthcare costs may limit coverage of or lower reimbursement for the procedures associated with the use of our product candidates once approved and, as a result, they may not cover or provide adequate payment for our product candidates. The downward pressure on healthcare costs in general, particularly prescription drugs and biologics and surgical procedures and other treatments, has become intense. As a result, increasingly high barriers are being erected to the entry of new products. The cost containment measures that payors and providers are instituting and the effect of any healthcare reform initiative implemented in the future could impact our revenue from the sale of our products once approved.
We believe that there will continue to be proposals by legislators at both the federal and state levels, regulators and third-party payors to reduce costs and potentially affect individual healthcare benefits. Certain of these changes could impose additional limitations on the rates we will be able to charge for any future products or the amounts of reimbursement available for any future products from governmental agencies or third-party payors.
We are and will continue to be dependent on third parties for strategically important tasks, and our ability to develop our products, obtain regulatory approval or bring products to market may fail if these third parties do not perform as we expect.
We are subject to risks related to our reliance on third parties in that we will:
| • | seek to enter into collaboration arrangements to fund development and commercialization of our products; |
| • | rely on CROs to conduct key elements of research by which our products are developed; |
| • | rely on Contract Development Organizations (“ CDOs ”) to develop key components of our products; |
| • | retain individual contractors or contracting organizations to perform critical functions in our company, including functions associated with senior management positions; and |
| • | seek to enter into joint development agreements for the manufacture of both our RNA materials and human health products with partners outside the U.S. |
The activities performed by these third parties may be delayed or suspended in light of the
ongoing COVID-19 pandemic,
which may impact our ability to successfully develop and test our product candidates and research programs in a timely manner.Our reliance on these third parties for research and development activities will reduce our control over these activities but will not relieve us of our responsibilities. For example, we will remain responsible for ensuring that
67
each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the trial. Moreover, the FDA requires us to comply with standards, commonly referred to as Good Clinical Practices, or GCPs, for conducting, recording, and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity, and confidentiality of trial participants are protected. We also are required to register ongoing clinical trials and post the results of completed clinical trials on a government-sponsored database, ClinicalTrials.gov, within certain timeframes. Failure to comply with these responsibilities can result in fines, adverse publicity, and civil and criminal sanctions.
The FDA enforces GCP regulations through periodic inspections of clinical trial sponsors, principal investigators, and trial sites. If we or our CROs fail to comply with applicable GCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that, upon inspection, the FDA will determine that any of our future clinical trials will comply with GCPs. In addition, our clinical trials must be conducted with investigational medicines produced in accordance with the requirements in cGMP regulations. Our failure or the failure of our CROs to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process and could also subject us to enforcement action.
Communicating with outside parties can also potentially lead to mistakes as well as difficulties in coordinating activities. Furthermore, these third parties may also have relationships with other entities, some of which may be our competitors.
If any of these third parties, or others we have business relationships with, fail to meet their obligations to us it will increase our expenses, damage our reputation and could result in the delay or shutdown of the projects they support and result in our inability to bring some or all of our products to market.
Our collaboration arrangements may restrict or prevent our future business activity in certain markets or industries, which could harm our ability to grow our business.
We will seek to enter into collaborations by which, in exchange for funding of infrastructure, development or marketing of our products, we will grant to other parties exclusive rights to the development, production, marketing or distribution of selected products in specific geographies. For example, in March 2022 we granted Serum Institute of India Private Limited (“SIIPL”) an exclusive,
sub-licensable,
royalty-bearing license to use our proprietary technology platform to develop, manufacture and commercialize an mRNA shingles product and up to two other mRNA products in all territories other than the United States, the 27 member states of the European Union, the United Kingdom, Australia, Japan, New Zealand, Canada, South Korea, China, Hong Kong, Macau, and Taiwan. These rights may keep us from entering into alternative collaborations which may keep us from using capital effectively and limit our ability to grow our business. Any failure on the part of our collaboration partners to comply with applicable laws, regulatory requirements and/or applicable contractual obligations or to fulfill any responsibilities they may have to protect and enforce any intellectual property rights underlying product candidates could harm our reputation, including with regulatory authorities, have an adverse effect on revenues, as well as give rise to possible legal proceedings. Moreover, the failure or perceived failure by us to comply with our obligations under a collaboration agreement with any of our collaboration partners could cause the partner to make public statements against us or could result in litigation asserting that we have breached our obligations, any of which could have a material adverse effect on our reputation, business, financial condition, or results of operations.68
If we bring our human health products to market as planned, our relationships with customers and third-party payors will be subject to applicable anti-kickback, fraud and abuse and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, program exclusion, contractual damages, reputational harm and diminished profits and future earnings.
Healthcare providers, physicians and third-party payors play a primary role in the recommendation of any product for which we obtain marketing licensure or approval. Our future arrangements with third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute products for which we obtain marketing approval. Efforts to ensure that our business arrangements with third parties comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines and exclusion from government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. If any of the physicians or other providers or entities with whom we expect to do business are found not to be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government-funded healthcare programs.
We are subject to significant regulation with respect to manufacturing our products. The manufacturing facilities on which we will rely may not continue to meet regulatory requirements and have limited capacity.
All entities involved in the preparation of vaccines and products for clinical studies or commercial sale, including Azzar Group or any other contract manufacturers we may use, are subject to extensive regulation. Components of a finished therapeutic product approved or licensed for commercial sale or used in late-stage clinical studies must be manufactured in accordance with applicable good manufacturing practice requirements, or GMP. These regulations govern manufacturing processes and procedures (including record keeping) and the implementation and operation of quality systems to control and assure the quality of investigational products and products approved or licensed for sale. Poor control of production processes can lead to the introduction of adventitious agents or other contaminants, or to inadvertent changes in the properties or stability of our product candidates that may not be detectable in final product testing. We or our contract manufacturers must supply all necessary documentation in support of a BLA on a timely basis and must adhere to the FDA’s good laboratory practices, or GLP, and GMP regulations enforced by the FDA through its facilities inspection program. Our facilities and quality systems and the facilities and quality systems of some or all our third-party contractors must pass
a pre-approval inspection
for compliance with the applicable regulations as a condition of regulatory licensure and approval of our product candidates. In addition, the regulatory authorities may, at any time, audit or inspect a manufacturing facility involved with the preparation of our product candidates or the associated quality systems for compliance with GMP and other regulations applicable to the activities being conducted. If these facilities do not passa pre-approval plant
inspection or do not have a GMP compliance status acceptable for the FDA, FDA approval or licensure of the products will not be granted.As product candidates proceed through preclinical studies to late-stage clinical trials towards potential approval and commercialization, various aspects of the development program, such as manufacturing methods and formulation, are commonly altered along the way in an effort to optimize processes and results. Such changes carry the risk that they will not achieve these intended objectives. Any of these changes could cause our product candidates to perform differently and affect the results of planned clinical trials or other future clinical trials conducted with the materials manufactured using altered processes. Such changes may also require additional testing, FDA notification or FDA approval. This could delay or prevent completion of clinical trials, require conducting bridging clinical trials or the repetition of one or more clinical trials, increase clinical trial costs, delay or prevent approval of our product candidates and jeopardize our ability to commence sales and generate revenue. The regulatory authorities also may, at any time following licensure or approval of a product
69
for sale, audit our manufacturing facilities or those of our third-party contractors. If any such inspection or audit identifies a failure to comply with applicable regulations or if a violation of our product specifications or applicable regulations occurs independent of such an inspection or audit, we or the relevant regulatory authority may require remedial measures that may be costly and/or time-consuming for us or a third-party to implement and that may include the temporary or permanent suspension of a clinical study or commercial sales or the temporary or permanent closure of a facility. Any such remedial measures imposed upon us or third parties with whom we contract could materially harm our business.
If we or any of our third-party manufacturers fail to maintain regulatory compliance with applicable GMP requirements, the FDA can impose regulatory sanctions including, among other things, refusal to approve a pending application for a new product or biologic product, or revocation of
a pre-existing approval.
As a result, our business, financial condition and results of operations may be materially harmed.Additionally, if supply from one approved manufacturer is interrupted, there could be a significant disruption in commercial supply. An alternative manufacturer would need to be qualified through a BLA supplement which could result in further delay. The regulatory agencies may also require additional studies if a new manufacturer is relied upon for commercial production. Switching manufacturers may involve substantial costs and is likely to result in a delay in our desired clinical and commercial timelines.
These factors could cause the delay of clinical studies, regulatory submissions, required approvals or commercialization of our product candidates, cause us to incur higher costs and prevent us from commercializing our products successfully. Furthermore, if our suppliers fail to meet contractual requirements, and we are unable to secure one or more replacement suppliers capable of production at a substantially equivalent cost, our clinical studies may be delayed, or we could lose potential revenue.
If we, or if our service providers or any third-party manufacturers, fail to comply with regulatory requirements, we or they could be subject to enforcement actions, which could adversely affect our ability to market and sell a product we develop in the future.
We have a limited ability to manufacture materials for our research programs and preclinical studies and we do not operate any significant manufacturing facilities. We primarily rely on third-party contract manufacturing organizations (“
CMOs
”) for the manufacture of our materials for preclinical and clinical studies and expect to continue to do so and for commercial supply of any product candidates that we develop and for which we or our collaborators obtain marketing approval. Additionally, the activities performed by our CMOs may be delayed or suspended in light of theongoing COVID-19 pandemic,
which may impact our ability to successfully develop and test our product candidates, including in clinical trials, and research programs in a timely manner.Reliance on third-party manufacturers entails additional risks, including: the possible breach of the manufacturing agreement by the third party; the possible termination or nonrenewal of the agreement by the third party at a time that is costly or inconvenient for us; and reliance on the third party for regulatory compliance, quality assurance, safety, and pharmacovigilance and related reporting. Third-party manufacturers may not be able to comply with cGMP regulations or similar regulatory requirements outside the United States.
If we, or if our service providers or any third-party manufacturers, fail to comply with applicable federal, state or foreign laws or regulations, we could be subject to enforcement actions, which could adversely affect our ability to successfully develop, market and sell a product we develop in the future and could harm our reputation. These enforcement actions may include:
| • | restrictions on, or prohibitions against, marketing; |
| • | restrictions on importation; |
| • | suspension of review or refusal to approve new or pending applications; |
70
| • | suspension or withdrawal of product approvals; |
| • | product seizures or recalls; |
| • | operating restrictions; |
| • | injunctions; and |
| • | civil and criminal penalties and fines. |
Risks related to creating a new class of mRNA products
Relatively few mRNA-based therapeutic product candidates have been tested in animals or humans, and the data underlying the feasibility of developing mRNA-based therapeutic products is both preliminary and limited. We have not yet succeeded and may not succeed in demonstrating the efficacy and safety of any of our product candidates in clinical trials or in obtaining marketing approval thereafter. We have not yet completed a clinical trial of any product candidate and we have not yet assessed safety of any product candidate in humans. As such, there may be adverse effects from treatment with any of our current or future product candidates that we cannot predict at this time.
Other than the approvals ofand the Moderna Spikevax
the Pfizer-BioNTech COVID-19 Vaccine
COVID-19
Vaccine, and the previous Emergency Use Authorizations forthese COVID-19 vaccines,
no mRNA medicines have been granted EUA or have been granted full approval or licensure to date by the FDA or other regulatory agencies. Moreover, it is possible that FDA will decline to accept new EUA submissionsfor COVID-19 vaccine
candidates if it determines that the underlying health emergency no longer exists or warrants such authorization. Successful discovery and development of other mRNA medicines by us or our strategic collaborators is highly uncertain and depends on numerous factors, many of which are beyond our or their control. We have made and will continue to make a series of business decisions and take calculated risks to advance our development efforts and pipeline, including those related to mRNA technology, delivery technology, and manufacturing processes, which may be shown to be incorrect based on further work by us, our strategic collaborators, or others. Our products that appear promising in the early phases of development may fail to advance, experience delays in the clinic, experience clinical holds, or fail to reach the market for many reasons, including:| • | discovery efforts at identifying potential mRNA medicines may not be successful; |
| • | nonclinical or preclinical study results may show potential mRNA medicines to be less effective than desired or to have harmful or problematic side effects; |
| • | clinical trial results may show potential mRNA medicines to be less effective than expected (e.g., a clinical trial could fail to meet one or more endpoint(s)) or to have unacceptable side effects or toxicities; |
| • | adverse effects in any one of our clinical programs or adverse effects relating to our mRNA, or our lipid nanoparticles (“LNPs”), may lead to delays in or termination of one or more of our programs; |
| • | the insufficient ability of translational models to reduce risk or predict outcomes in humans, particularly given that each component of investigational medicines and development candidates may have a dependent or independent effect on safety, tolerability, and efficacy, which may, among other things, be species-dependent; |
| • | manufacturing failures or insufficient supply of cGMP materials for clinical trials, or higher than expected cost could delay or set back clinical trials, or make mRNA-based medicines commercially unattractive; |
| • | our improvements in the manufacturing processes for this new class of medicines and potential medicines may not be sufficient to satisfy the clinical or commercial demand of our investigational medicines or regulatory requirements for clinical trials; |
71
| • | changes that we make to optimize our manufacturing, testing or formulating of cGMP (current good manufacturing process regulations as enforced by the FDA) materials could impact the safety, tolerability, and efficacy of our investigational medicines and development candidates; |
| • | pricing or reimbursement issues or other factors may delay clinical trials or make any mRNA medicine uneconomical or noncompetitive with other therapies; |
| • | failure to timely advance our programs or receive the necessary regulatory approvals or a delay in receiving such approvals, due to, among other reasons, slow or failure to complete enrollment in clinical trials, withdrawal by trial participants from trials, failure to achieve trial endpoints, additional time requirements for data analysis, data integrity issues, preparation of a BLA, or the equivalent application, discussions with the FDA or EMA, a regulatory request for additional nonclinical or clinical data, or safety formulation or manufacturing issues may lead to our inability to obtain sufficient funding; and |
| • | the proprietary rights of others and their competing products and technologies that may prevent our mRNA medicines from being commercialized. |
Currently, mRNA is considered a gene therapy product by the FDA. Unlike certain gene therapies that irreversibly alter cell DNA and could act as a source of side effects, mRNA-based medicines are designed to not irreversibly change cell DNA; however, side effects observed in gene therapy could negatively impact the perception of mRNA medicines despite the differences in mechanism. In addition, because no product in which mRNA is the primary active ingredient has been approved without first being authorized for emergency use, the regulatory pathway for approval is uncertain. The number and design of the clinical trials and preclinical studies required for the approval of these types of medicines have not been established, may be different from those required for gene therapy products, or may require safety testing like gene therapy products. Moreover, the length of time necessary to complete clinical trials and to submit an application for marketing approval for a final decision by a regulatory authority varies significantly from one pharmaceutical product to the next, and may be difficult to predict.
Adverse events in clinical trials of our investigational medicines or in clinical trials of others developing similar products and the resulting publicity, as well as any other adverse events in the field of mRNA medicine, or other products that are perceived to be similar to mRNA medicines, such as those related to gene therapy or gene editing, could result in a decrease in the perceived benefit of one or more of our programs, increased regulatory scrutiny, decreased confidence by patients and clinical trial collaborators in our investigational medicines, and less demand for any product that we may develop. In addition, responses by U.S., state, or foreign governments to negative public perception may result in new legislation or regulations that could limit our ability to develop any investigational medicines or commercialize any approved products, obtain or maintain regulatory approval, or otherwise achieve profitability. More restrictive statutory regimes, government regulations, or negative public opinion would have an adverse effect on our business, financial condition, results of operations, and prospects and may delay or impair the development of our investigational medicines and commercialization of any approved products or demand for any products we may develop.
Risks Related to Our Plant Health Program
Our inability to obtain regulatory approvals, or to comply with ongoing and changing regulatory requirements, could delay or prevent sales of the products we are developing and commercializing.
The field testing, manufacture, sale and use of crop protection, plant health and plant nutrition products are extensively regulated by the EPA and other state, local and foreign governmental authorities. These regulations substantially increase the time and cost associated with bringing our products to market. If we do not receive the necessary governmental approvals to test, manufacture and market our products, or if regulatory authorities revoke our approvals, do not grant approvals in a timely manner or grant approvals subject to restrictions on their use, we may be unable to sell our products in the United States or other jurisdictions, which could result in a reduction in our future revenues.
72
As we introduce new formulations of and applications for our products, we may need to seek EPA approval prior to commercial sale. For any such approval, the EPA may require us to fulfill certain conditions within a specified period of time following initial approval. We will also be required to obtain regulatory approval from other state and foreign regulatory authorities before we market our products in their jurisdictions, some of which have taken, and may take, longer than anticipated.
Some of these states and foreign countries may apply different criteria than the EPA in their approval processes. Although federal pesticide law preempts separate state and local pesticide registration requirements to some extent, state and local governments retain authority to control pesticide use within their borders.
There can be no assurance that we will be able to obtain regulatory approval for marketing our additional products or new product formulations and applications we are developing. Although the EPA has in place a registration procedure for biopesticides there can be no assurance that all of our products or product extensions will be eligible for this streamlined procedure or that additional requirements will not be mandated by the EPA that could make the procedure more time consuming and costly for our future products.
Additionally, for certain state registration and registration in jurisdictions outside of the United States, all products need to be proven efficacious for each proposed crop-pest combination, which can require costly field trial testing, and a favorable result is not assured. Because many of the products that may be sold by us must be registered with one or more government agencies, the registration process can be time consuming and expensive, and there is no guarantee that the product will obtain all required registrations. We may seek registration in some jurisdictions and not in others. California is one of the largest and most important producers of agricultural products in the world. As such, we view California as one of the most natural and attractive markets for our products, but it is also very stringent in its regulations, generally requiring more time and effort, and lacking legally mandated deadlines for its reviews of reduced-risk biopesticides. Therefore, gaining concurrent approvals with the EPA, other states and other countries may not always be achievable. Even if we obtain all necessary regulatory approvals to market and sell our products, they will be subject to continuing review and extensive regulatory requirements, including
periodic re-registrations. The
EPA, as well as state and foreign regulatory authorities, could withdraw a previously approved product from the market upon receipt of newly discovered information, including an inability to comply with their regulatory requirements or the occurrence of unanticipated problems with our products, or for other reasons.If our field trials are unsuccessful, we may be unable to obtain regulatory approval of, or commercialize, our products on a timely basis.
The successful completion of multiple field trials in domestic and foreign locations on various crops is critical to the success of our product development and marketing efforts. If our ongoing or future field trials are unsuccessful or produce inconsistent results or unanticipated adverse side effects on crops or
on non-target organisms,
or if we are unable to collect reliable data, regulatory approval of our products could be delayed, or we may be unable to commercialize our products. In addition, more than one growing or treatment season may be required to collect sufficient data and we may need to collect data from different geographies to prove performance for customer adoption. Although we have conducted many successful field trials we cannot be certain that additional field trials conducted on a greater number of acres, or on crops for which we have not yet conducted field trials, will be successful. Moreover, the results of our ongoing and future field trials are subject to a number of conditions beyond our control, including weather-related events such as drought or floods, severe heat or frost, hail, tornadoes and hurricanes, or low or no natural occurrence of the pests intended for testing. Generally, we pay third parties, such as growers, consultants and universities to conduct field tests on our behalf. Incompatible crop treatment practices or misapplication of our products by these third parties or lack of sufficient occurrence of the identified pests in nature for a particular trial could impair the success of our field trials.73
Crop protection products must be extensively tested for safety, efficacy and environmental impact before they can be registered for production, use, sale or commercialization in a given market.
The regulatory approvals process is lengthy, costly, complex and in some markets unpredictable, with requirements that can vary by product, technology, industry and country. The regulatory approvals process for products that incorporate novel modes of action or new technologies can be particularly unpredictable and uncertain due to the then-current state of regulatory guidelines and objectives, as well as governmental policy considerations
and non-governmental organization
and other stakeholder considerations. In certain jurisdictions, we will need to periodically renew any regulatory approvals which may require us to demonstrate compliance with shifting or more stringent requirements as time passes.The markets for biological agricultural products are intensely competitive, rapidly changing and undergoing consolidation. We may be unable to compete successfully against our current and future competitors, which may result in price reductions, reduced margins and the inability to achieve market acceptance for our products.
Many entities with significantly greater resources than us are engaged in developing biological agricultural products, including BASF SE, Valent BioSciences Corporation, Corteva Agriscience, UPL Limited, and FMC Corporation. Each of these competitors is a major multinational agrichemical company with longer operating histories, significantly greater resources, greater brand recognition, established global sales channels and a larger base of customers than we have. As a result, they may be able to devote greater resources to the manufacture, promotion or sale of their products, receive greater resources and support from independent distributors, initiate or withstand substantial price competition, offer full-line discounts we cannot match, or more readily take advantage of acquisition or other opportunities. Further, many of the large agrichemical companies have a more diversified product offering, which may give these companies an advantage in meeting customers’ needs by enabling them to offer a broader range of crop protection, plant nutrition and plant health products. In addition, we could face competition in the future from new,
well-financed start-up companies.
Customers in the agricultural sector tend to be loyal to major brands and distributors and are generally cautious in adopting new products and technologies, making the barriers to entry high in this market. If new products or technologies fail to achieve immediate results, they may never achieve significant customer adoption and, even if immediate and positive results are achieved, adoption may take several growing seasons as multi-year purchasing agreements expire and product-specific equipment is replaced.
Products incorporating biotechnology-derived traits and crop protection products must be extensively tested for safety, efficacy and environmental impact before they can be registered for production, use, sale or commercialization in a given market. In certain jurisdictions, we must periodically renew our approvals for both biotechnology and crop protection products, which typically require us to demonstrate compliance with then-current standards which generally are more stringent since the prior registration. The regulatory approvals process is lengthy, costly, complex and in some markets unpredictable, with requirements that can vary by product, technology, industry and country. The regulatory approvals process for products that incorporate novel modes of action or new technologies can be particularly unpredictable and uncertain due to the then-current state of regulatory guidelines and objectives, as well as governmental policy considerations
and non-governmental organization
and other stakeholder considerations.Changes in the regulatory environment could adversely impact our ability to produce and/or sell certain products in domestic and foreign markets or could increase the cost of doing so.
Changes in the regulatory environment, particularly in the U.S., Brazil, China, India, Argentina and the European Union, could adversely impact our ability to produce and/or sell certain products in domestic and foreign markets or could increase the cost of doing so. Additionally, changes to the regulatory environment may be influenced
by non-government public
pressure as a result of negative perception regarding the use of our crop74
protection products. We are sensitive to this regulatory risk given the need to obtain and maintain pesticide registrations in every country in which we sell our products. Many countries
require re-registration of
pesticides to meet new and more challenging requirements; while we defend our products vigorously, thesere-registration
processes may result in significant additional data costs, reduced number of permitted product uses, or potential product cancellation. Compliance with changing laws and regulations may involve significant costs or capital expenditures or require changes in business practice that could result in reduced profitability. In the European Union, the regulatory risk specifically includes the chemicals regulation known as REACH (Registration, Evaluation, and Authorization of Chemicals), which requires manufacturers to verify through a special registration system that their chemicals can be marketed safely, as well as Regulation (EC) No 1107/2009, governing plant protection products, which sets forth rules for the authorization, sale, use, and control of plant protection products.Customers have historically perceived biological agricultural products as more expensive and less effective than conventional products. To succeed, we will need to continue to change that perception. To the extent that the market for biological agricultural products does not further develop or customers elect to continue to purchase and rely on conventional chemical products, our market opportunity will be limited.
Any decline in U.S. agricultural production could have a material adverse effect on the market for biopesticides and on our results of operations and financial position.
Conditions in the U.S. agricultural industry will significantly impact demand for our products. The U.S. agricultural industry has contracted in recent periods, and can be affected by a number of factors, including weather patterns and field conditions, current and projected grain inventories and prices, domestic and international demand for U.S. agricultural products and U.S. and foreign policies regarding trade in agricultural products. State and federal governmental policies, including farm subsidies and commodity support programs, as well as the prices of fertilizer products and the prices at which produce may be sold, may also directly or indirectly influence the number of acres planted, the mix of crops planted and the use of pesticides for particular agricultural applications.
Sales of our plant health products will depend upon weather conditions, seasonal variation and other factors
We expect to commercially launch Calantha, our RNAi Colorado potato beetle product in 2022, assuming we are able to obtain EPA and state approvals according to our current plans, which assumes EPA approval by the end of the second quarter of 2022. If and when we do begin selling our product to farmers, our sales will be subject to weather conditions and other factors beyond our control, which may cause our operating results to fluctuate significantly quarterly and annually.
Weather conditions, natural disasters and other factors affect planting and growing seasons and incidence of pests and plant disease, and accordingly affect decisions by our distributors, direct customers and end users about the types and amounts of pest management and plant health products to purchase and the timing of use of such products. In addition, disruptions that cause delays by growers in harvesting or planting can result in the movement of orders to a future quarter, which would negatively affect the quarter and cause fluctuations in our operating results. Customers also may purchase large quantities of our products in a particular quarter to store and use over long periods of time or time their purchases to manage their inventories, which may cause significant fluctuations in our operating results for a particular quarter or year.
Our activities are subject to extensive federal, state, local and foreign governmental regulations. These regulations may prevent us or our collaborators from developing or commercializing products in a timely manner or under technically or commercially feasible conditions and may impose expenses, delays and other impediments to our product development and registration efforts.
In the United States, the EPA regulates
our bio-based pest
management products under the Federal Food, Drug and Cosmetics Act (“FFDCA
”), the Food Quality Protection Act (“FQPA
”) and the Federal Insecticide,75
Fungicide and Rodenticide Act (“
FIFRA
”). In addition, some of our plant health products are regulated as fertilizers, auxiliary plant substances, soil amendments, beneficial substances and/or biostimulants in each of the fifty states.In general, FIFRA prohibits the sale or distribution of any pesticide, a product category that includes the insecticides, fungicides, and acaricides we are developing, unless that pesticide is registered with the EPA. To register a pesticide with the EPA, the applicant must demonstrate that the product will not cause unreasonable adverse effects on human health or the environment. These adverse effects include any unreasonable risk to man or the environment, taking into account the economic, social, and environmental costs and benefits of the use of the pesticide, as well as any human dietary risk from residues that result from use of the pesticide in or on any food consistent with the FFDCA. In the course of its evaluation of a pesticide, EPA assesses the impact that a pesticide may have on endangered species
and non-target organisms.
In order to commercialize a product for the U.S. agricultural market, we must complete specified toxicology studies, submit a registration dossier to the EPA demonstrating that the product does not pose unreasonable risks to human health or the environment, respond adequately to any deficiencies identified by the EPA through its risk assessment process and obtain the EPA’s approval of our labeling. The EPA must also establish a tolerance level for the product or issue a tolerance exemption. We must separately obtain any applicable state or foreign regulatory approvals. Moreover, because our products contain
novel RNA-based active
ingredients, there will generally be no previously registered pesticide product containing that active ingredient and, as a result, the use of each of our products will require a new registration under FIFRA and the establishment of a tolerance under Section 408 of the FFDCA or the issuance of a tolerance exemption.We have not previously obtained any EPA approvals for our biopesticides, and it is uncertain whether the EPA will approve any of our products or whether it will place conditions of approval that adversely impact our ability to sell them. Although the EPA has evaluated and approved other companies’ RNA products before, our products may differ materially from the products that have previously received approval, and the lack of precedent makes it more difficult to predict whether or when the EPA will grant approval or the conditions that it might impose on approval.
Even if our biopesticide product candidates are approved by the EPA, as with any pesticide, they would continue to be subject to review by the EPA and state regulatory agencies. The EPA has the authority to revoke the registration or impose limitations on the use of any of our pest management products if we do not comply with the regulatory requirements, if unexpected problems occur with a product or if the EPA receives other newly discovered adverse information. Our inability to obtain regulatory approvals, or to comply with ongoing and changing regulatory requirements, could delay or prevent sales of the products we are developing and commercializing.
Inadequate funding, staffing or shutdowns of the government agencies that regulate us could prevent new products and services from being developed or commercialized in a timely manner or otherwise prevent those agencies from performing normal business functions on which the operation of our business may rely, which could negatively impact our business.
Our research and development activities are also subject to federal, state and local worker safety, air pollution, water pollution and solid and hazardous waste regulatory programs, ongoing compliance requirements, permitting requirements, and periodic inspection. Any significant noncompliance could impact our ability to operate. In addition, any future expansion of our manufacturing capabilities may require additional or expanded permitting, and such permitting requirements may impede or prevent our ability to operate.
76
Our agricultural products may fail to meet the criteria for desirable certifications such
as “non-GMO” or
“organic” and may cause the plants or products to which they are applied also to lose these certifications, reducing the addressable market for and value of our products.The use of products created through synthetic biology processes is generally prohibited in organic food supply chains and the U.S. Department of Agriculture and similar regulators outside the U.S. will not permit an “organic” certification unless the supply chain from field to table is free of such products. We do not currently expect any of our agricultural products to qualify for “organic” certifications, which will keep us from selling into a market with potentially higher returns and which will limit the size of the addressable market for which our products can be used. In addition, the standards associated with certifications such
as “non-GMO” and
“organic” can differ significantly between countries and jurisdictions within countries (such as states and cities) and, even when these standards are clearly established, the application of the standards for certification may differ depending on the third-party organizations conducting verification.Genetically modified products are currently subject to public debate and heightened regulatory scrutiny, either of which could prevent or delay the adoption of our products. Claims that genetically modified products are unsafe or pose a danger to the environment may influence public attitudes and lead to our product not gaining public acceptance. The subject of genetically modified organisms has received negative publicity in the United States and particularly in Europe, and such publicity has aroused public debate. The adverse publicity in Europe could lead to greater regulation and trade restrictions.
We may have product liability claims if our agricultural products damage individuals or property and may need to recall items which do or could cause such damage.
Our agricultural products are intended to be used to improve yield in the human food supply chain. If our products are used for an application they are not intended for, become adulterated or mislabeled we may need to recall such products. A widespread product recall could result in significant losses due to the costs of a recall, the destruction of product inventory, and lost sales due to the unavailability of product for a period of time. We could also suffer losses from a significant product liability judgment against us. A significant product recall or product liability case could also result in adverse publicity, damage to our reputation, and a loss of confidence in our products, which could have an adverse effect on our business, results of operations and financial condition and the value of our brands.
Risks Related to our Animal Health Program
We currently have one animal health product which is intended to control the Varroa destructor mite, a honeybee parasite; however, the honeybee ecosystem is complex and it difficult to measure the overall efficacy of this product since there are multiple factors other than Varroa mites contributing to the decline in honeybee populations.
The one animal health product in our product pipeline, which we call GS15, is intended to support the health of honeybees by using dsRNA to discourage the spread of the Varroa destructor mite, a honeybee parasite. A multitude of stressors can contribute to declines in honeybee health, making it difficult to determine whether or the degree to which GS15 benefits honeybees and, by implication, beekeepers. Other factors that impact honeybee health include pesticides, environmental stressors, inadequate nutrition, parasites, pathogens, and poor management practices. No one factor has been identified as the primary cause of honeybee health decline or “bee colony collapse,” including the Varroa destructor mite parasite which GS15 is designed to control. Determining dominating stressors to honeybee health is challenging to characterize and pinpoint. To complicate matters, many of the factors contributing to honeybee health decline interact, making it difficult in some circumstances to identify dominant factors. Unless we are able to develop clear correlations between GS15 use and specific successful outcomes in beehives, GS15 (assuming it obtains regularly approval) may not have strong or any commercial prospects.
77
Delivering the active ingredient in our bee health product effectively to Varroa mites is challenging.
Until we complete our field trials, it is unclear whether we will be able to deliver our product first to bees and, through bees, to Varroa mites, in a manner that will effectively impede mite function. Challenges in active ingredient delivery may interfere with the effectiveness of our product.
When testing our Varroa mite product under the high-dose conditions commonly required by the EPA for pesticide approval, we have observed a dose response in bees and an increase in bee mortality.
Although our product is intended to impact Varroa mites and not bees, it may have unanticipated impacts on bee health. The EPA typically seeks a 10x dose safety factor in order to approve a product for commercial use. We tested our product at this concentration and observed significant bee mortality. At the concentrations we would expect under normal conditions, however, our recent field trials demonstrated no negative effects. Our most recent data indicates that bee mortality at the 10x dose safety factor likely results from the extremely high viscosity of RNA when administered at that concentration, which prevents bees from feeding. If there is a significant relationship between our product and bee mortality, that relationship may undercut our product’s intended function of protecting bees and may impair our ability to obtain regulatory approval of our product.
The EPA will evaluate our Varroa mite product without a precedent product, which may result in the need to conduct additional field trials and lengthen the regulatory review period. If we cannot reduce bee mortality experienced in high-dose safety factor testing, the EPA may not approve our product or may impose labeling requirements that materially limit the commercial attractiveness of the product.
Our Varroa mite product is subject to EPA approval under FIFRA. Although the EPA has evaluated and approved RNA products before, our product may differ materially from previously approved products. After we perform our planned field trials and studies, the EPA may request further studies and data to effectively evaluate and approve our product. We anticipate that this process will require additional time to complete and may delay regulatory approval and commercialization. Moreover, other markets, such as those in the European Union, may require additional data or information prior to granting approval, and they may impose more stringent conditions on any approval.
One challenge we face in securing EPA approval for our Varroa mite product is that EPA typically seeks to ensure that a product does not cause adverse effects even when administered at a dose equal to ten times the dose that bees and other organisms would likely receive in typical use. Ordinarily, pesticides are applied at high doses in the field to account for anticipated losses to the environment resulting from degradation, runoff and other factors, and these losses are anticipated to reach as high as 95%. As a result, in a conventional field application scenario, organisms are generally expected to receive the actual field use rate, even when the product is applied at ten times that rate. When we tested our Varroa mite product in the laboratory at the required level of ten times the field use rate, the higher concentration of the product caused the treated bee food to become highly viscous, which limited consumption and resulted in bee starvation. We did not observe these adverse effects either when our product was administered at the field use rate or when our product was administered at the high-dose rate in the field. Because our product is delivered inthrough
a ready-to-use formulation
a pre-measured pouch
delivery system, rather than through conventional spraying, we do not believe that our product presents a material risk that bees will be exposed to concentrations greater than the field use rate. We are negotiating with the EPA to modify its customary 10x safety factor protocol for both beesand non-target organisms
to account for differences between the delivery system for our product and traditional field application methods. We may not be successful in these negotiations, and extended negotiations, even if ultimately successful, could delay regulatory approval and commercial introduction of the product. If the EPA does not modify its safety factor protocol for our Varroa mite product, we may be unable to obtain regulatory approval to commercialize the product in the United States, which would limit our growth opportunities. Even if the product receives EPA approval, it may receive labeling with warnings of potentially harmful effects on bees or other organisms, which would materially limit the commercial attractiveness of the product to potential customers.78
We purchased some of the intellectual property related to our RNA honeybee product from Bayer Crop Science, a subsidiary of Bayer, which now owns Monsanto. It is well known that Monsanto has had significant pushback from environmental groups regarding its technology and practices, and our product may be hard to market since it was purchased from Bayer.
Despite rigorous testing during
the pre-commercialization phase,
if GS15 comes to market, there may be opponents of our RNA technology or synthetic biology generally who raise concerns about the potential for adverse effects of our products on human or animal health, plants and the environment. Because GS15 in part originated with Monsanto, the many negative public perceptions associated with Monsanto could impair our ability to bring GS15 to market and can affect the timing of, and whether we are able to obtain, government approvals for our products.Even after approvals are granted for GS15, public concern may lead to increased regulation or legislation or litigation against government regulators concerning prior regulatory approvals, which could affect our sales and results of operations, and which may adversely affect sales of GS15 to beekeepers, including due to their concerns about available markets for the sale of crops or other products including those derived from biotechnology. Genetically modified products are currently subject to public debate and heightened regulatory scrutiny, either of which could prevent or delay the adoption of GS15. Claims that genetically modified products are unsafe or pose a danger to the environment may influence public attitudes and lead to our product not gaining public acceptance. The subject of genetically modified organisms has received negative publicity in the United States and particularly in Europe, and such publicity has aroused public debate. The adverse publicity in Europe could lead to greater regulation and trade restrictions.
In addition, opponents of agricultural synthetic biology have attacked facilities used by agricultural biotechnology companies, and may launch future attacks against beekeepers’ hives and our field testing sites and research, production, or other facilities, which could affect our sales and our costs.
The research and development process for GS15 is expensive with little immediate return, and the field trials associated with honeybees in general are susceptible to circumstances outside of our control.
Although our field trial operators are under agreement not to extract honey from their hives during field trials, there is the risk that honey could be extracted impermissibly and find their way into the commercial market thereby impairing our ability to meet regulatory requirements or obtain regulatory approval.
Furthermore, beekeepers tend to be migratory as they serve the needs of seasonal crops and the environments in which the hives are placed can vary. These variables introduce the risk that hives could be damaged or otherwise compromised so as to require their removal from the field trials or field trial results which make it difficult for us to accurately measure the effects of GS15.
Beekeeping practices and results also vary and are subject to factors outside of our control. For example, the overall health and productivity of a beehive is dependent on the queen, how she is mated, how well the nurse bees are taking care of her and larvae, and how well forager bees are able to bring back food to the hive. One hive may have bees that go north and a hive right next to it may have bees that go south to look for food, which causes variability in food sources and potentially in test results. This variability in testing can make it difficult or impossible for us to accurately isolate the effects of GS15 which may in turn increase the cost of field testing, the length and likelihood of regulatory approval and the commercial viability of the GS15 product.
Our GS15 product is intended to be used in commercial beehives and used in a fashion which will expose the product only to bees and the Varroa destructor mite. If GS15 is used inappropriately and is consumed by invertebrates other than the Varroa destructor mite, it could be harmful to those invertebrates.
According to a September 2020 report published by the Environmental Directorate of the Organization for Economic Cooperation and Development (“OECD”) entitled
Considerations
for the Environmental Risk Assessment
79
of the Application of Sprayed or Externally Applied
ds-RNA-Based
Pesticides
It may be difficult to convince beekeepers to adopt our product, and to use it in the way prescribed for maximum effect.
Although we foresee that our product will be effective in controlling mites that impact bees, beekeepers may ultimately perceive shortcomings in treatment efficacy. This may result in reduced demand for our product or the selection of other treatment options.
Additionally, we will not be able to control how beekeepers will ultimately use our product, and misuse may result in reduced product efficacy, and thus reduced demand. For example, if beekeepers were to dilute our product formulation before application, the diluted product might leave hives subject to microbial contamination and allow microbes to consume our product, impeding its ability to affect mite function.
GS15 is susceptible to purity risks associated with scaling up manufacturing and we are also developing our own process for manufacturing our product at scale.
Commercial production of our product candidates will involve quantities several orders of magnitude higher than our current level of production. We may face challenges producing a product that meets applicable purity specifications at that scale, and may encounter other issues related to scaled up manufacturing.
While Bayer Crop Science (from which we obtained some intellectual property for the product) had its own proprietary methods for manufacturing, we did not license these methods, and we are developing our own methods of manufacturing GS15. We are currently developing a way to make this product with our cell-free platform so that it is economical to produce. Uncertainties related to this platform may ultimately limit our ability to produce the product.
Our product may require approval from other federal and state regulatory bodies.
As discussed above, state regulatory approvals may also be required for our product, which may delay commercialization. In addition, the EPA may not be the only federal agency with jurisdiction over products designed to eliminate honeybee pests. In 2017, the FDA, EPA, and USDA released a document entitled “Modernizing the Regulatory System for Biotechnology Products: Final Version of the 2017 Update to the Coordinated Framework for the Regulation of Biotechnology.” That document outlines the role that the three agencies have in biotechnology approvals. While it is clear that EPA has jurisdiction over pesticide and insecticide products pursuant to FIFRA, USDA also asserts jurisdiction over honeybees in some circumstances. As a result, we may need USDA evaluation and approval of our product, in addition to other unanticipated regulatory approvals. These additional approvals may delay commercialization.
Risks Related to Intellectual Property
If we are unable to obtain and maintain sufficient intellectual property protection for our products, platform, methods, and technology, or if the scope of the intellectual property protection obtained is not sufficiently broad, competitors could develop and commercialize products similar or identical to ours, and our ability to successfully commercialize our products may be impaired.
Our commercial success depends in part on our ability to protect our intellectual property and proprietary technologies. We rely on a combination of patent, trade secret and trademark laws, and nondisclosure,
80
confidentiality and other contractual restrictions to protect our intellectual property and proprietary technology. However, these legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep any competitive advantage. If we fail to obtain, maintain and protect our intellectual property, third parties may be able to compete more effectively against us and our competitive position could be adversely affected, as could our business, financial condition, results of operations and prospects. In addition, we may incur substantial costs related to litigation or other patent proceedings in our attempts to recover or restrict use of our intellectual property.
To the extent that our intellectual property offers inadequate protection, or is found to be invalid or unenforceable, we would be exposed to a greater risk of direct competition. If our intellectual property does not provide adequate coverage of our competitors’ products and methods, our competitive position could be adversely affected, as could our business, financial condition, results of operations and prospects. Both the patent application process and the process of managing patent and other intellectual property disputes are generally unpredictable, time-consuming and expensive.
Our success depends in large part on our own and any future licensor’s ability to obtain and maintain protection of the intellectual property we may own or license, whether solely or jointly, particularly patents, in the United States and other countries with respect to our products, platform, methods, and technologies. We apply for patents to protect our products, platform, methods, technologies and commercial activities, as we deem appropriate. However, obtaining and enforcing patents is costly, time-consuming and complex, and we may fail to apply for patents on important products, methods, and technologies in a timely fashion or at all, or we may fail to apply for patents in potentially relevant jurisdictions. Moreover, we may fail to obtain issuance of any of the patent applications that we do file. Publications of discoveries in scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing, or in some cases not at all. Therefore, we cannot know with certainty whether we were the first to make the inventions claimed in our patents or pending patent applications, or whether we were the first to file for patent protection of such inventions.
We may not be able to file and prosecute all necessary or desirable patent applications, or maintain, enforce and license any patents that may issue from such patent applications, at a reasonable cost or in a timely manner or in all jurisdictions. It is also possible that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection. Moreover, we may not develop additional proprietary products, methods and technologies that are patentable. Even if we believe an innovation to be patentable and file patent applications, the United States Patent Office (“
USPTO
”) and other patent offices may not find our innovations to be patentable and may refuse to grant patent rights. We may not have the right to control the preparation, filing and prosecution of patent applications, or to maintain the rights to patents which may be licensed from or to third parties or held jointly with third parties. In connection with any future licensing arrangements with third parties, these patents and applications may not be prosecuted and enforced by such third parties in a manner consistent with the best interests of our business. We currently own and may in the future own patents, patent applications, and other intellectual property jointly with third parties. In certain jurisdictions, including in the United States, joint owners of a patent are free to license rights under the patent to third parties without any compensation to or permissionfrom co-owners. If
we are unable to negotiate licenses on commercially reasonable termswith co-owners of
patents, in order to exclusively control commercial licensing or commercial use ofour co-owned patents
or if agreements allowing us such control are found unenforceable,then co-owners may
be able to license to our competitors and other third parties without our permission and without compensation to us. Failure to control exclusive rights under intellectual property as discussed above may have an adverse effect on our competitive position, business, financial condition, and results of operations.81
We may become involved in lawsuits to enforce our intellectual property or defend against third-party claims of infringement, misappropriation, or other violations of intellectual property which could be expensive, time consuming, and unsuccessful and may prevent or delay development and commercialization efforts, and could harm our competitive position and business prospects.
Litigation may be necessary for us to enforce our patent and proprietary rights, defend against intellectual property claims brought by third parties and/or determine the scope, coverage and validity of third parties’ intellectual property rights. Litigation on these matters has been prevalent in our industries and we expect that this will continue. To determine the priority of inventions, we may have to initiate and participate in interference proceedings declared by the U.S. Patent and Trademark Office that could result in substantial legal fees and could substantially affect the scope of our patent protection. Also, our intellectual property may be subject to significant litigation and administrative proceedings such as invalidity, unenforceability, IPR,
re-examination,
opposition, or other post-grant proceedings against our patents. The outcome of any litigation or other proceeding is inherently uncertain and might not be favorable to us and we might not be able to obtain licenses to technology that we require or a competitor may have already obtained an exclusive license to such technology in the relevant fields. Even if such licenses are obtainable, they may not be available at a reasonable cost. We could therefore incur substantial costs related to royalty payments for licenses obtained from third parties, which could negatively affect our competitive position, business, financial condition, or results of operations and/or make it unfeasible to commercialize a given product. In some cases, the outcome of litigation may be to enjoin us from commercializing or using a technology protected by third-party intellectual property. We could encounter delays in product introductions, or interruptions in product sales, as we develop alternative methods or products or we may need to cease sales of a product altogether if we are unable to develop alternatives that avoid the relevant third-party intellectual property.If we resort to legal proceedings to enforce our intellectual property rights or to determine the validity, scope and coverage of the intellectual property or other proprietary rights of others, the proceedings could be burdensome, time-consuming, and expensive, even if we were to prevail. Moreover, it may not be possible for us to enforce jointly owned patents in the U.S. or other jurisdictions without the cooperation of other owners.
Our commercial success may depend in part on
our non-infringement of
the patents or proprietary rights of third parties and/or the invalidity or unenforceability of such patent or proprietary rights of others. Numerous significant intellectual property issues have been litigated, and will likely continue to be litigated, between or among existing and new participants in our relevant markets and competitors may assert that our products or methods infringe their intellectual property rights as part of a business strategy to impede the successful entry into or continued presence in those markets. Third parties may assert that we are employing their proprietary technology without authorization. For example, numerous third-party patents exist in fields relevant to the company’s business and planned products, such as biologics, mRNA vaccines and therapies, and RNA interference (“RNAi
”) for crop protection. Our competitors and others have patents and may in the future obtain patents and may claim that use, manufacture, sale, or importation of our products infringe these patents. Moreover, as we move into new markets and applications for our technologies, incumbent participants in such markets may assert their patents and other proprietary rights against us as a means of slowing or preventing our entry into such markets, or as a means to extract substantial license and royalty payments from us.We have received notice in the past that our proposed products or methods may require an intellectual property license from others in order to be developed, produced, used, or sold. In each instance, we have reviewed the underlying intellectual property and either negotiated for a license or determined that no license was necessary. For future notices, if we are unable to successfully negotiate licenses or determine that no license is necessary, we may be unable to bring impacted products to market. Moreover, allegations that we violate third-party intellectual property could lead to disputes, including litigation.
The outcome of litigation is uncertain and even if we believe that we do not violate asserted third-party intellectual property or that such intellectual property is invalid or unenforceable or otherwise not legally
82
protectable, our defense may be unsuccessful. If a court were to find that we have violated the intellectual property rights of a third party, an injunction and/or an award of damages may have an adverse effect on our business, financial condition, and results of operations. Remedies for intellectual property infringement or misappropriation may include an injunction against future sales of products or use of methods, statutory damages, enhanced damages, punitive damages, attorney’s fees, an award of lost profits and/or a reasonable royalty and prejudgment interest. Damages may in some cases exceed our own profits on sales found to be infringing. Even if we are successful in defending claims, defending intellectual property litigation, particularly patent or trade secret litigation, can be prohibitively burdensome and expensive.
We may be required in the future to license patent rights from third-party owners in order to develop or continue to sell or use a product or method. If we cannot obtain such licenses, or if such owners do not properly maintain or enforce the patents underlying such licenses, our competitive position and business prospects will be harmed.
We develop products, platform, and methods in technological areas and industries that are critical to public health and agriculture—areas in which there is considerable competition. A third-party may hold intellectual property rights, including patent rights and trade secrets that are important or necessary to the development, manufacture, or commercialization of our current or future product candidates. It may be necessary for us to use the patented or proprietary technology of one or more third parties to manufacture or commercialize our product candidates, in which case we would be required to obtain a license from these third parties on commercially reasonable terms, or our business could be harmed. If any such patents were to be asserted against us, there is no assurance that a court would find in our favor or that, if we choose to or are required to seek a license, that a license to any of these patents would be available to us on acceptable terms or at all.
To the extent that we enter into any patent licenses with third parties, if such third parties fail to properly maintain the licensed patents or if those patents are found to be invalid or unenforceable, then we could be subject to additional competition due to the loss of exclusive
or non-exclusive rights.
Defending and protecting intellectual property rights in foreign jurisdictions is costly and sometimes prohibitively expensive.
Obtaining patent protection in every country is prohibitively expensive and, as we attempt to choose jurisdictions for intellectual property protection, we may fail to protect our intellectual property in relevant jurisdictions where we do business, and thereby cause a loss of revenue and profits or other impacts on our ability to manufacture and export our products.
Competitors may use our proprietary technologies in jurisdictions where we have not obtained patent protection to manufacture or develop their own products. Such competitors may export otherwise infringing products, or products made through otherwise infringing methods, to territories where we may have or obtain patent protection, but where patent enforcement is not as strong as in the United States or where no protection is available for products made abroad through methods that infringe a local patent. These products may also compete with our products in jurisdictions where we do not have any issued or licensed patents and any future patent claims or other intellectual property rights may not be effective or sufficient to prevent them from so competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of some countries do not favor the enforcement of patents and other intellectual property protection, which could make it difficult for us to stop the infringement of our patents or trademarks or the misappropriation of our trade secrets generally. Proceedings to enforce our intellectual property rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not
83
prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful.
Many countries, including India, Japan, China, and some European nations have compulsory licensing laws under which a patent owner may be compelled under specified circumstances (including a matter of public policy) to grant licenses to third parties. In those countries, we may have limited remedies if patents are infringed or if we are compelled to grant a license to a third-party, which could diminish the value of those patents. This could limit our potential revenue opportunities. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
The United States has the absolute right to manufacture and use patented inventions and to allow others to manufacture and use patent inventions for the United States, for reasonable compensation.
Pursuant to 28 U.S.C. § 1498, the United States government has the absolute right to use or manufacture any patented inventions for reasonable compensation. No injunction of patent infringement is available against the United States and damages are limited to reasonable compensation only. Moreover, a patent owner may not obtain damages or an injunction against a private entity for its infringing use or manufacture for the United States. Such suits may only be brought against the United States and only for reasonable compensation. In the past the United States has relied on such rights to use or manufacture inventions from third parties other than the patentee. In the future, the United States may rely on its rights to infringe our current and future patents or to allow others to make or use our inventions on behalf of the United States. If the United States were to use, or allow others to use for the United States, our patented technology, platform, methods, or products, then we would only be entitled to reasonable compensation and would not be entitled to an injunction to prevent infringement. As with all intellectual property litigation, proceedings against the United States for patent infringement could be burdensome, time-consuming, and expensive. Even if we were to prevail on a patent infringement action against the United States, any remedy would not likely compensate us for the full extent of the financial harm from such infringement due to the limited remedies available against the United States under the law. Such infringement could adversely affect our competitive position, business, financial condition, results of operations, and prospects.
When inventions are developed with government funding, the United States retains
a paid-up license
to such inventions and may compel us to grant licenses to third parties for little or no compensation.Under 35 U.S.C. § 200, when inventions arise, even in part, through use of public funds, the United States may retain an. In such cases, the United States would have the right to use, or allow others to use on its behalf, our inventions, whether protected by patent or trade secret, without any compensation to us.
et seq.
irrevocable, paid-up license
to practice or have practiced on its behalf, such inventions, whether protected by patent or trade secret. That is, the United States has the absolute right to use, and allow others to use on its behalf, such intellectual property without any compensation to the holder of the intellectual property. We have acquired and developed and may in the future acquire or develop trade secrets or obtain patents on inventions developed, in full or in part, with funding from the United States government. A court could also find that one or more of our current patents or applications are covered by 35 U.S.C. § 200 et seq
For inventions subject to 35 U.S.C. § 200, the United States also
et seq.
retains march-in rights.
These march-in rights
apply in certain situations, including, for example, when action is necessary to alleviate public health or safety needs or when an inventor is not taking reasonable steps to make its technology useful to the public. In such situations, the United States has the right to compel the innovator to grant licenses to third parties. If such a finding were made with respect to our current or future patents or trade secrets, then we may not be able to prevent competitors from practicing our patented inventions and/or may be compelled to license our competitors to practice our inventions for little or no monetary compensation. Any of the foregoing events could have an adverse effect on our business, financial condition and results of operations.84
Confidentiality agreements with employees and others may not adequately prevent disclosure of trade secrets and other proprietary information.
We currently rely, and intend to rely in the future, on trade
secrets, know-how and
technology that are not protected by patents to maintain our competitive position. In order to protect our proprietary technology and processes, we also rely in part on confidentiality agreements with our collaborators, employees, consultants, and sponsored researchers and other advisors. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. These agreements may be found by a court to be unenforceable or invalid. We may fail to enforce our agreements in Court if we are compelled to present them as evidence but are unable to locate and provide copies. Moreover, when employees with knowledge of our trade secrets and confidential information leave us and join new employers, it may be difficult or impossible for us to detect or prove misappropriation of our confidential information and trade secrets by the former employee and/or the former employee’s new employer. In addition, others may independently discover trade secrets and proprietary information, and in such cases, we could not assert any trade secret rights against such party. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could adversely affect our competitive position, business, financial condition and results of operations.Changes in patent laws or patent jurisprudence could diminish the value of patents in general, thereby impairing our ability to protect our products and platforms.
The patent position of biotechnology, life sciences, and pharmaceutical companies generally is highly uncertain, involves complex legal and factual questions and has in recent years been the subject of much litigation. In addition, the laws of foreign countries may not protect our rights to the same extent as the laws of the United States. For example, European patent law restricts the patentability of methods of treatment of the human body more than United States law does. In another example, some jurisdictions prevent the patenting of certain biotechnology inventions outside of narrow coverage for exact nucleotide sequences.
As a result, the issuance, scope, validity, enforceability and commercial value of our patent rights are highly uncertain. Our pending and future patent applications may not result in patents being issued which protect our technology, platform, methods, or products, in whole or in part, or which effectively prevent others from commercializing competitive products. Our issued patents may be found to be invalid or unenforceable in a post grant proceeding before patent offices or in patent litigation before courts in the United States or other countries. Changes in either the patent laws or interpretation of the patent laws in the United States and other countries may diminish the value of our patents, narrow the scope of our patent protection, or result in the invalidity or unenforceability of our patents.
Various courts, including the U.S. Supreme Court, have recently rendered decisions that impact the scope of patentability of certain inventions or discoveries relating to our technology and commercial goals. Specifically, these decisions have substantially increased the probability that patent claims will be ruled patent ineligible for reciting a natural phenomenon, law of nature or abstract idea. Furthermore, in view of these decisions, the USPTO has published and continues to publish revised guidelines for patent examiners to apply when examining claims for patent eligibility. Patent claims relating to software algorithms, biologically-derived compositions, methods for analyzing biological systems and other subject matters that underlie our technology and commercial goals are impacted by these changes.
On September 16, 2011, the Leahy-Smith America Invents Act (the “
Leahy-Smith Act
”) was signed into law. The Leahy-Smith Act made a number of significant changes to United States patent laws. These include provisions that affect the way patent applications are prosecuted and challenged at the USPTO and may also affect patent litigation. The USPTO has developed and continues to develop regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with85
the Leahy-Smith Act, and in particular, the first to file provisions, became effective on March 16, 2013. Accordingly, it is not clear what, if any, impact the Leahy-Smith Act, subsequent rulemaking, and judicial interpretation of the Leahy-Smith Act and regulations will have on the operation of our business in the future. The Leahy-Smith Act and its continued implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement and/or defense of our issued patents, all of which could have an adverse effect on our business and financial condition.
Actions taken by the U.S. Congress, federal courts and USPTO have from time to time narrowed the scope of patent protection available in certain circumstances and weakened the rights of patent owners in certain situations. Similar changes have been made by authorities in other jurisdictions. In addition to increasing uncertainty with regard to the ability to obtain patents in the future, such changes create uncertainty with respect to the value of patents, once obtained. Depending on decisions by authorities in various jurisdictions, the laws and regulations governing patents could change in unpredictable ways that may have an adverse effect on our ability to obtain new patents and to defend and enforce our existing patents and patents that we might obtain in the future, harming our business, competitive position, financial condition, results of operations, and prospects.
Patent reform legislation could increase uncertainties and costs surrounding the prosecution of our patent applications and the enforcement, validity, or defense of our issued patents. There has been recent public discussion around loosening of patent protection for inventions important to addressingSuch reforms or similar changes in connection with future pandemics or other public health emergencies, could have an adverse effect on any patent protection on our products and methods.
the SARS-CoV-2 pandemic.
We cannot assure you that our patent portfolio will not be negatively impacted by the current uncertain state of the law, new court rulings or changes in guidance or procedures issued by governments or patent offices around the world. From time to time, the U.S. Supreme Court, other federal courts, the U.S. Congress or the USPTO may change the standards of patentability, scope and validity of patents within the biotechnology, life sciences, and other relevant technologies and any such changes, or any similar adverse changes in the patent laws of other jurisdictions, could have a negative impact on the our business, financial condition, prospects and results of operations.
Moreover, we may be subject to a
third-party pre-issuance submission
of prior art to the USPTO, or become involved in opposition, derivation, reexamination, inter partes review, post-grant review or interference proceedings challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our patent rights, allow third parties to commercialize our technology or products or use our proprietary methods and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize products without infringing third-party patent rights.Our current or future issued patents could be found invalid or unenforceable if challenged or could be construed narrowly such that they do not cover our products or methods or those used by our competitors.
It is possible that none of our current or future pending patent applications will result in issued patents in a timely fashion or at all. Even if our patent applications issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors from competing with us or otherwise provide us with any competitive advantage. Moreover, we cannot predict the breadth of claims that may be allowed or enforced in our patents. Our issued patents may not be construed to cover our own or our competitors’ products or methods. Our competitors may be able to circumvent or design around our patents by developing similar or alternative technologies or products in
a non-infringing manner.
In addition, if the breadth or strength of protection provided by our patents and patent applications is threatened, it could dissuade companies from collaborating with us to license, develop or commercialize current or future product candidates.The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our patents may be challenged in the courts or patent offices in the United States and abroad. Such challenges may
86
result in loss of exclusivity, in patent claims being narrowed or invalidated, or in patents being held unenforceable which could limit our ability to stop others from using or commercializing similar or identical technology, methods, and products, or limit the duration of the patent protection of our technology, methods, and products.
The inventorship and/or ownership rights for our patents and patent applications may be challenged by third parties. Such challenges could result in invalidation of such patents, the loss of ownership of such patents, or loss of exclusive rights to such patents, which could result in increased competition and could limit or eliminate our ability to stop others from using or commercializing similar or identical technology, methods, and products or require us to obtain a license from third parties on commercially reasonable terms to secure exclusive rights, or our business could be harmed. If any such challenges to inventorship and/or ownership were asserted, there is no assurance that a court would find in our favor or that, if we choose to seek a license, such license would be available to us on acceptable terms or at all.
Patent terms may be inadequate to protect our competitive position for an adequate amount of time.
Patents have a limited lifespan. In the United States, the natural expiration of a patent is generally 20 years after the
first non-provisional filing
date. In the United States, a patent’s term may, in certain cases, be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the USPTO in examining and granting a patent, or in certain cases, by patent term extension for patents covering certain pharmaceutical products requiring regulatory approval. In the United States a patent’s term also may be shortened if a patent is terminally disclaimed over a commonly owned patent or a patent naming a common inventor and having an earlier expiration date. The life of a patent, and the protection it affords, is limited. Therefore, even if patents covering our products and methods are obtained, once the patent life has expired, for our current or future platform, products, methods or technologies, we may be open to competition, including, for example, from biosimilar or generic versions of our products. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.Obtaining and maintaining patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated
for non-compliance with
these requirements.The USPTO and European and other patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. In addition, periodic maintenance, renewal and annuity fees on any issued patent are due to be paid to the USPTO and European and other patent agencies over the lifetime of a patent. While an inadvertent failure to make payment of such fees or to comply with such provisions can in many cases be cured by additional payment of a late fee or by other means in accordance with the applicable rules, there are situations in which such noncompliance will result in the abandonment or lapse of the patent or patent application, and the partial or complete loss of patent rights in the relevant
jurisdiction. Non-compliance events
that could result in abandonment or lapse of a patent or patent application include failure to respond to official actions within prescribed timelimits, non-payment of
fees and failure to properly legalize and submit formal documents within prescribed time limits. If we or our licensors fail to maintain the patents and patent applications covering our platform, technology, methods, or products or if we or our licensors otherwise allow patents or patent applications to be abandoned or lapse, our competitors might be able to enter the market, which would hurt our competitive position and could impair our ability to successfully commercialize our product candidates, which could have an adverse effect on our business and financial condition.87
We may become subject to claims for ownership of intellectual property, payment, or royalties for assigned invention rights by our employees, contractors, and collaborators, which could result in litigation and adversely affect our business.
We may be subject to claims that former employees, collaborators or other third parties have an interest in, or right to compensation, with respect to our current patent and patent applications, future patents and patent applications, or other intellectual property as an inventor
or co-inventor. For
example, we may have inventorship or ownership disputes from consultants, former employees, or others who are involved in developing a product for us. Litigation may be necessary to defend against these and other claims challenging inventorship or ownership or claiming the right to compensation. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as ownership of, exclusive ownership of, or the right to use and/or exclude others from using, valuable intellectual property. Such an outcome could have an adverse effect on our business, financial condition, results of operations, and prospects. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.We generally enterwith our employees pursuant to which such individuals assign to us all rights to any inventions created in the scope of their employment or engagement with us. If compelled to provide copies of relevant agreements as evidence of such arrangements, we may not be able to locate and provide copies of such agreements and may therefore be unable to assert such agreements. Moreover, such agreements could be found to be invalid or unenforceable. Although our employees have agreed to assign to us invention rights and have specifically waived their right to receive any special remuneration for such assignment beyond their regular salary and benefits, we may face claims demanding ownership of inventions or remuneration in consideration for assigned inventions.
into assignment-of-invention agreements
We may not be able to protect and enforce our trademarks and trade names, or build name recognition in our markets of interest or may be subject to claims of trademark infringement thereby harming our competitive position.
We have filed, and may continue in the future to file trademark applications to protect certain of our intellectual property; however, we cannot guarantee that we will be successful in registering our trademarks. Our registered or unregistered trademarks or trade names may be challenged, infringed, circumvented, declared generic, lapsed or determined to be infringing on or dilutive of other marks. We may not be able to protect our rights in these trademarks and trade names, which we need in order to build name recognition. In addition, third parties have filed, and may in the future file, for registration of trademarks that may impede our ability to build brand identity and possibly leading to market confusion. Third parties have identified potential conflicts between their marks and our marks that may arise in the future. In addition, there could be potential trade name or trademark infringement claims brought by owners of trademarks or trademarks that incorporate variations of our trademarks or trade names. Such claims may require us to cease use of our trademarks or change our company or product names. Further, we may in the future enter into agreements with owners of such third-party trade names or trademarks to avoid potential trademark litigation which may limit our ability to use our trade names or trademarks in certain fields or territories. Over the long term, if we are unable to establish name recognition based on our trademarks and trade names, then we may not be able to compete effectively, and our business, financial condition, results of operations and prospects may be adversely affected. Our efforts to enforce or protect our proprietary rights related to trademarks, domain names, or other intellectual property may be ineffective and could result in substantial costs and diversion of resources. Any of the foregoing events could have an adverse effect on our business, financial condition and results of operations.
88
Intellectual property rights do not necessarily address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business, or permit us to maintain our competitive advantage. The following examples are illustrative:
| • | Others may be able to develop or make products, platform, methods or technology that are similar to products, platform, methods or technology we have developed or will develop, but that are not covered by the claims of the patents that we own or have licensed and are not protectable through trade secret law. |
| • | We or our licensors or strategic partners might not have been the first to make the inventions covered by the issued patent or pending patent application that we own or have exclusively licensed, and therefore our patents may be found to be invalid or our patent applications may be rejected. |
| �� | • | We or our licensors or strategic partners might not have been the first to file patent applications covering certain of our inventions, and therefore our patents may be found to be invalid or our patent applications may be rejected. |
| • | Others may independently develop or make similar or alternative products, platform, methods or technology or duplicate any of our products, platform, methods or technology without infringing our intellectual property rights. For example, independent development of such products, platform, methods or technology would make it impossible for us to assert trade secret rights against such third parties. If such third parties publish the details of such independently developed products, platform, methods or technology, then we could lose any trade secret protection even as against others. |
| • | It is possible that our pending patent applications will not lead to issued patents. |
| • | Issued patents that we own or have exclusively licensed may not provide us with any competitive advantages, or may be held invalid or unenforceable, as a result of legal challenges by our competitors. |
| • | Our competitors might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets. |
| • | Our competitors may use our manufacturing methods to produce products in jurisdictions in which we do not have patent protection on our manufacturing methods and may export such products for sale other jurisdictions, including our major commercial markets for us. Patents on such methods in our major commercial markets may not protect against such product sales. |
| • | We may not develop additional proprietary technologies that are patentable or protectable through other intellectual property rights. |
| • | The intellectual property rights of others may have an adverse effect on our business. |
Should any of these events occur, they could significantly harm our business, competitive position, financial condition, results of operations and prospects.
Risks Related to Ownership of New GreenLight Common Stock
An active trading market for the New GreenLight Common Stock may never develop or be sustained.
We cannot assure you that an active trading market for the New GreenLight Common Stock will develop on the Nasdaq or elsewhere. If an active trading market does not develop, or develops but is not maintained, you may have difficulty selling any shares of New GreenLight Common Stock due to the limited public float. Accordingly, we cannot assure you of your ability to sell your shares of New GreenLight Common Stock when desired or the prices that you may obtain for your shares.
89
The market price of the New GreenLight Common Stock may be volatile, which could result in substantial losses for investors.
The market price of the New GreenLight Common Stock may fluctuate due to a variety of factors, including:
| • | the need to obtain regulatory approval for our product candidates; |
| • | the risk that clinical trials will not demonstrate that our therapeutic product candidates are safe and effective; |
| • | the risk that our product candidates will have adverse side effects or other unintended consequences, which could impair their marketability; |
| • | the risk that our product candidates do not satisfy other legal and regulatory requirements for marketability in one or more jurisdictions; |
| • | the risks of enhanced regulatory scrutiny of RNA-based products, including mRNA and dsRNA; |
| • | the potential inability to achieve our goals regarding scalability, affordability and speed of commercialization of our product candidates; |
| • | the anticipated need for additional capital to achieve our business goals; |
| • | changes in the industries in which we operate; changes in laws and regulations affecting our business, |
| • | the potential inability to implement or achieve business plans, forecasts, and other expectations after the completion of the proposed transaction; |
| • | actual or anticipated fluctuations in our operating results, including fluctuations in our quarterly and annual results; |
| • | operating expenses being more than anticipated; |
| • | the failure or discontinuation of any of our product development and research programs; |
| • | the success of existing or new competitive businesses or technologies; |
| • | announcements about new research programs or products of our competitors; |
| • | developments or disputes concerning patent applications, issued patents or other proprietary rights; |
| • | the recruitment or departure of key personnel; |
| • | litigation and governmental investigations involving us, our industry or both; |
| • | investor perceptions of us or our industry; |
| • | negative perceptions of publicly traded companies that have gone public through business combinations with publicly traded special purpose acquisition companies; |
| • | sales of New GreenLight Common Stock by us or by our insiders or other stockholders; |
| • | the expiration of market standoff or lock-up agreements; |
| • | general economic, industry and market conditions; and |
| • | the COVID-19 pandemic, natural disasters or major catastrophic events. |
Recently, stock markets in general, and the market for life sciences technology companies in particular, have experienced significant price and volume fluctuations that have often been unrelated or disproportionate to changes in the operating performance of the companies whose stock is experiencing those price and volume fluctuations, particularly in light of the
current COVID-19 pandemic.
Broad market and industry factors may seriously affect the market price of New GreenLight Common Stock, regardless of our actual operating90
performance. These fluctuations may be even more pronounced in the trading market for New GreenLight Common Stock. Following periods of such volatility in the market price of a company’s securities, securities class action litigation has often been brought against that company. Because of the potential volatility of the price of New GreenLight Common Stock, we may become the target of securities litigation in the future. Securities litigation could result in substantial costs and divert management’s attention and resources from our business.
Our failure to meet the continued listing requirements of Nasdaq could result in a delisting of our securities.
If we fail to satisfy the continued listing requirements of Nasdaq, such as the corporate governance requirements or the minimum closing bid price requirement, Nasdaq may take steps to delist our securities. Such a delisting would likely have a negative effect on the price of our securities and would impair your ability to sell or purchase our securities when you wish to do so. In the event of a delisting, we can provide no assurance that any action taken by us to restore compliance with listing requirements would allow our securities to become listed again, stabilize the market price or improve the liquidity of our securities, prevent our securities from dropping below the Nasdaq minimum bid price requirement or prevent
future non-compliance with
Nasdaq’s listing requirements. Additionally, if our securities are not listed on, or become delisted from, Nasdaq for any reason, and are quoted on the OTC Bulletin Board, an inter-dealer automated quotation system for equity securities that is not a national securities exchange, our ability to issue addition securities or obtain additional financing in the future, the analyst coverage, and the liquidity and price of our securities may be more limited than if we were quoted or listed on Nasdaq or another national securities exchange. As a result, you may be unable to sell your securities unless a market can be established or sustained.The exercise of registration rights may adversely affect the market price of the New GreenLight Common Stock.
Certain of our stockholders have registration rights for certain securities. Pursuant to the Subscription Agreements for the PIPE Financing and the Investor Rights Agreement, we have registered a substantial number of shares of New GreenLight Common Stock for resale and for issuance upon exercise of the Public Warrants. We are obligated to file one or more resale “shelf” registration statements to register such securities, use commercially reasonable efforts to cause such registration statements to be declared effective by the SEC within specified periods, and keep such registration statements effective for up to three years thereafter. We are also obligated to file other registration statements, including for underwritten offerings of New GreenLight Common Stock, in specified circumstances. Sales of a substantial number of shares of New GreenLight Common Stock pursuant to these registration statements in the public market could occur at any time. These sales, or the perception in the market that the holders of a large number of shares intend to sell shares, could reduce the market price of the New GreenLight Common Stock. For more information relating to the registration rights under the Subscription Agreements and the Investor Rights Agreements, see Exhibit 4.3, “.”
Description of Securities—Registration Rights
If securities or industry analysts do not publish or cease publishing research or reports about New GreenLight, our business, or our market, or if they change their recommendations regarding our securities adversely, the price and trading volume of our securities could decline.
The trading market for our securities will be influenced by the research and reports that industry or securities analysts may publish about us, our business, markets or competitors. Securities and industry analysts do not currently, and may never, publish research on New GreenLight. If no securities or industry analysts commence coverage of us, our share price and trading volume would likely be negatively impacted.
As a former shell company, we will face certain disadvantages relative to companies that pursue a traditional initial public offering, including ineligibility for certain forms and rules for extended periods.
ENVI was a special purpose acquisition company, or SPAC, a form of shell company under the rules of the SEC. Shell companies are more highly regulated
than non-shell operating
companies and face significant91
additional restrictions on their activities under federal securities laws. As a result of the Business Combination, New GreenLight ceased to be a shell company. However, companies that were formerly shell companies continue to face disadvantages under SEC rules, including (a) the inability to use
Form S-3 until
at least one year after the filing of information equivalent to that required by Form 10 after ceasing to be a shell company, (b) the inability to qualify as a “well-known seasoned issuer” and file automatically effective registration statements for three years after ceasing to be a shell company, (c) the inability to “incorporate by reference” information in certain registration statements filed under the Securities Act for a period of three years after ceasing to be a shell company, (d) the inability to use most free writing prospectuses until at least three years after a qualifying business combination, (e) the inability to useForm S-8 to
register shares issuable in connection with certain compensatory plans and arrangements until 60 days after the filing of information equivalent to that required by Form 10, (f) the inability of stockholders to rely on Rule 144 for resales of securities until at least one year after the filing of information equivalent to that required by Form 10 and the provision of current public information, and (g) exclusion from certain safe harbors for offering-related communications under the Securities Act for three years after ceasing to be a shell company, including for research reports and certain communications in connection with business combinations. We expect that these disadvantages will make it more challenging and expensive, and create greater risks and delays, for both us and our stockholders to offer securities. These challenges may make our securities less attractive than those of companies that are not former shell companies and may raise our relative cost of capital.Reports published by analysts, including projections in those reports that differ from our actual results, could adversely affect the price and trading volume of New GreenLight Common Stock.
Securities research analysts may establish and publish their own periodic projections for New GreenLight. These projections may vary widely and may not accurately predict the results we actually achieve. The share price of New GreenLight Common Stock may decline if our actual results do not match the projections of these securities research analysts. If any of the analysts who may cover New GreenLight issue an adverse or misleading opinion regarding New GreenLight, our business model, our intellectual property or our stock performance, change their recommendation regarding shares of New GreenLight Common Stock adversely, provide relatively more favorable recommendations about our competitors or if the clinical trials and operating results fail to meet the expectations of analysts, the price of shares of New GreenLight Common Stock would likely decline. If one or more of these analysts ceases coverage of New GreenLight or fails to publish reports on New GreenLight regularly, the share price or trading volume of New GreenLight Common Stock could decline. If no analysts commence coverage of New GreenLight, the market price and volume for the New GreenLight Common Stock could be adversely affected.
A significant portion of the outstanding shares of New GreenLight Common Stock are restricted from immediate resale by certain
temporary lock-up arrangements
but may be sold into the market in the near future. This could cause the market price of New GreenLight Common Stock to drop significantly, even if our business is doing well.Substantially all of the shares of New GreenLight Common Stock issued in the Merger are subject to certain and.
temporary lock-up arrangements,
which will expire no later than 180 days after the consummation of the Business Combination. Upon the expiration ofthose lock-up arrangements,
and assuming the continued availability of a prospectus for resales of the shares covered by the registration statement on FormS-1
that we initially filed with the Commission on February 7, 2022 (as amended from time to time), substantially all of the outstanding shares of New GreenLight Common Stock will be freely tradable. Accordingly, sales of a substantial number of shares of New GreenLight Common Stock in the public market could occur at any time. These sales, or the perception in the market that the holders of a large number of shares intend to sell shares, could reduce the market price of the New GreenLight Common Stock. For more information relating tothe lock-up arrangements,
please see Exhibit 4.3,“Description of Securities –
Lock-Up”
“Description of Securities – Registration Rights – Investor Rights Agreement.”
92
We have broad discretion in the use of the net proceeds from the exercise of outstanding public and private warrants, if any, and may not use them effectively.
As of March 15, 2022, we had outstanding Public Warrants to purchase 10,350,000 shares of New GreenLight Common Stock and Private Placement Warrants to purchase 2,062,500 shares of New GreenLight Common Stock, in each case for an exercise price of $11.50 per share. The warrants may never be exercised for cash, in which case we will not receive any proceeds from such exercise. Because of this uncertainty, we have no specific plans for the proceeds that we may receive from warrant exercises, if any, and we intend to use any such proceeds for general corporate purposes and working capital, including the funding of our clinical programs. Our management will have broad discretion in the application of the net proceeds. Our management may spend a portion or all of the net proceeds in ways that our stockholders may not desire or that may not yield a favorable return. The failure by our management to apply these funds effectively could harm our business, financial condition, results of operations and prospects. Pending their use, we may invest the net proceeds from the exercise of such warrants, if any, in a manner that does not produce income or that loses value.
We do not expect to pay any dividends for the foreseeable future. Investors may never obtain a return on their investment.
You should not rely on an investment in New GreenLight Common Stock to provide dividend income. We do not anticipate that we will pay any dividends to holders of New GreenLight Common Stock in the foreseeable future. Instead, we plan to retain any earnings to maintain and expand our existing operations, fund our research and development programs and continue to invest in our commercial infrastructure. In addition, any future credit facility or financing we obtain may contain terms prohibiting or limiting the amount of dividends that may be declared or paid on New GreenLight Common Stock. Accordingly, investors must rely on sales of their shares after price appreciation, which may never occur, as the only way to realize any return on their investment. As a result, investors seeking cash dividends should not purchase New GreenLight Common Stock.
The Charter designates the Delaware Court of Chancery as the exclusive forum for specified disputes between New GreenLight and our stockholders and also provides that the federal district courts are the exclusive forum for resolving any complaint asserting a cause of action arising under the Securities Act, each of which could limit the ability of our stockholders to choose the judicial forum for disputes with New GreenLight or our directors, officers or employees.
Our Charter provides that, unless we consent in writing to the selection of an alternative forum (an “
Alternative Forum Consent
”), to the fullest extent permitted by the applicable law, the Court of Chancery of the State of Delaware will be the sole and exclusive forum for any stockholder (including a beneficial owner) to bring (i) any derivative action or proceeding brought on behalf of New GreenLight, (ii) any action asserting a claim of breach of a fiduciary duty owed by any director, officer or other employee of New GreenLight to us or our stockholders, (iii) any action asserting a claim against New GreenLight, our directors, officers or employees arising pursuant to any provision of the DGCL, the Charter or the Bylaws, or (iv) any action asserting a claim against New GreenLight, our directors, officers or employees governed by the internal affairs doctrine, subject to specified exceptions. This provision will not apply to suits brought to enforce a duty or liability created by the Securities Exchange Act of 1934, as amended, for which claims may be brought in any U.S. federal court, or any other claim for which the federal courts have exclusive jurisdiction.To prevent having to litigate claims in multiple jurisdictions and the threat of inconsistent or contrary rulings by different courts, among other considerations, the Charter also provides that, unless we give an Alternative Forum Consent, the federal district courts of the United States will, to the fullest extent permitted by law, be the exclusive forum for the resolution of any complaint asserting a cause of action arising under the Securities Act or the rules and regulations promulgated thereunder. However, Section 22 of the Securities Act creates concurrent jurisdiction for federal and state courts over all such actions. Accordingly, both state and federal courts have jurisdiction to entertain such claims, and there is uncertainty whether a court would enforce the Charter’s choice of forum provision applicable to Securities Act claims.
93
Any person or entity purchasing or otherwise acquiring any interest in any of our securities shall be deemed to have notice of and consented to the foregoing charter provisions. Although we believe these exclusive forum provisions benefit us by providing increased consistency in the application of Delaware law and federal securities laws in the types of lawsuits to which each applies, the exclusive forum provisions may limit a stockholder’s ability to bring a claim in a judicial forum of its choosing for disputes with New GreenLight or any of our directors, officers, or other employees, which may discourage lawsuits with respect to such claims against New GreenLight and our current and former directors, officers, or other employees. In addition, a stockholder that is unable to bring a claim in the judicial forum of its choosing may be required to incur additional costs in the pursuit of actions which are subject to the exclusive forum provisions described above. Our stockholders will not be deemed to have waived our compliance with the federal securities laws and the rules and regulations thereunder as a result of our exclusive forum provisions. Further, in the event a court finds either exclusive forum provision contained in the Charter to be unenforceable or inapplicable in an action, we may incur additional costs associated with resolving such action in other jurisdictions, which could harm our results of operations.
Delaware law and provisions in our Charter and Bylaws might discourage, delay or prevent a change in control of our company or changes in our management and, therefore, depress the trading price of the New GreenLight Common Stock.
Our status as a Delaware corporation and the anti-takeover provisions of the Delaware General Corporation Law may discourage, delay or prevent a change in control by prohibiting us from engaging in a business combination with an interested stockholder for a period of three years after the person becomes an interested stockholder without the approval of holders of 66
2
/3
% of the voting power of our stockholders other than the interested stockholder, even if a change of control would be beneficial to our existing stockholders. In addition, our Charter and Bylaws contain provisions that may make the acquisition of our company more difficult, including the following:| • | Our board of directors is classified into three classes of directors with staggered three-year terms, and directors can only be removed from office for cause by the affirmative vote of holders of a majority of the voting power of our then-outstanding capital stock; |
| • | certain amendments to our Charter will require the approval of stockholders holding three-fourths of the voting power of our then-outstanding capital stock; |
| • | any stockholder-proposed amendment to the Bylaws that is not recommended by the New GreenLight Board will require the approval of stockholders holding three-fourths of the voting power of our then-outstanding capital stock; |
| • | our stockholders are only able to take action at a meeting of stockholders and cannot take action by written consent for any matter; |
| • | vacancies on our board of directors can be filled only by our board of directors and not by stockholders; |
| • | only the New GreenLight Board, pursuant to a written resolution adopted by a majority of the New GreenLight Board, is authorized to call a special meeting of stockholders; |
| • | certain litigation against New GreenLight can only be brought in Delaware; |
| • | the Charter authorizes undesignated preferred stock, the terms of which may be established by the New GreenLight Board, which shares may be issued without the approval of the holders of our capital stock; and |
| • | advance notice procedures apply for stockholders to nominate candidates for election as directors or to bring matters before an annual meeting of stockholders. |
These anti-takeover defenses could discourage, delay, or prevent a transaction involving a change in control of New GreenLight. These provisions could also discourage proxy contests and make it more difficult for
94
stockholders to elect directors of their choosing or to cause us to take other corporate actions they desire, any of which, under certain circumstances, could limit the opportunity for our stockholders to receive a premium for their shares of our capital stock.
The ability to use our net operating losses to offset future taxable income may be subject to numerous limitations.
As of December 31, 2021, GreenLight had U.S. federal and state net operating loss carryforwards, or NOLs, of $232.1 million and $197.4 million, respectively. If not utilized, the federal NOLs generated before 2018 of approximately $27.1 million will expire at various dates through 2037 and the state NOLs will expire at various dates through 2040. The federal NOLs generated after 2017 of approximately $205.0 million have an indefinite carryforward period. GreenLight or New GreenLight may potentially use these U.S. federal and state NOLs to offset against taxable income for U.S. federal and state income tax purposes. However, the use of these NOLs may be subject to numerous limitations under the U.S. Internal Revenue Code of 1986, as amended, or the Code, and under state tax laws. Among such limitations, Section 382 of the Code may limit the use of these NOLs in any year for U.S. federal income tax purposes in the event of certain past or future changes in ownership of GreenLight or New GreenLight. An ownership change under Section 382 of the Code, referred to in this discussion as an ownership change, generally occurs if one or more stockholders or groups of stockholders who own at least 5% of a company’s stock increase their ownership by more than 50 percentage points over their lowest ownership percentage within a rolling three-year period. An ownership change in respect of New GreenLight also could be deemed to be an ownership change in respect of GreenLight. We have not conducted a Section 382 study to determine whether the use of our NOLs is impaired under Section 382 of the Code as a result of any prior ownership change. GreenLight may have previously undergone one or more ownership changes. In addition, the Business Combination and PIPE Financing, or future issuances or sales of our stock, including certain transactions involving our stock that are outside of our control, could result in future ownership changes. Ownership changes that have occurred in the past or that may occur in the future, including in connection with the Business Combination and PIPE Financing, could result in the imposition of an annual limit under Section 382 of the Code on the amount
of pre-ownership change
NOLs and other tax attributes that GreenLight or New GreenLight can use to reduce its taxable income, potentially increasing or accelerating its liability for income taxes, and also potentially causing those tax attributes to expire unused. States may impose similar limitations on the use of applicable NOLs. Any limitation on using NOLs, whether under Section 382 of the Code or otherwise under U.S. federal or state tax laws, could, depending on the extent of such limitation and the NOLs previously used, result in GreenLight or New GreenLight retaining less cash after payment of U.S. federal and state income taxes in respect of any year in which GreenLight or New GreenLight has taxable income, rather than losses, than GreenLight or New GreenLight would be entitled to retain if such NOLs were available as an offset against such income for U.S. federal and state income tax reporting purposes, which could adversely impact GreenLight or New GreenLight’s operating results.New GreenLight remains an “emerging growth company” and a “smaller reporting company”, and the reduced disclosure requirements applicable to emerging growth companies and smaller reporting companies may make the New GreenLight Common Stock less attractive to investors.
We are an “emerging growth company,” as defined in the JOBS Act. For so long as we remain an emerging growth company, we are permitted by SEC rules to, and plan to, rely on exemptions from certain disclosure requirements that are applicable to other SEC registered public companies that are not emerging growth companies. These exemptions include not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes Oxley Act, not being required to comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements, reduced disclosure obligations regarding executive compensation and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and any golden parachute payments not previously approved. As a result, the information we provide to stockholders will be different than the information that is
95
available with respect to other public companies that are not emerging growth companies. In this annual report, not all of the executive compensation-related information that would be required if we were not an emerging growth company has been included. If we were to continue to qualify as a “smaller reporting company,” as such term is defined in
Rule 12b-2 under
the Securities Exchange Act of 1934, after we cease to qualify as an emerging growth company, we would continue to be permitted to make certain reduced disclosures in our periodic reports and other documents that we file with the SEC. We expect to cease to qualify as a smaller reporting company before we cease to qualify as an emerging growth company. We cannot predict whether investors will find the New GreenLight Common Stock less attractive if we rely on these exemptions. If some investors find the New GreenLight Common Stock less attractive as a result, there may be a less active trading market for the New GreenLight Common Stock and our stock price may be more volatile.In addition, the JOBS Act provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards. This allows an emerging growth company to delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have elected to avail ourselves of this exemption from new or revised accounting standards and, therefore, we will not be subject to the same new or revised accounting standards as other public companies that have not made or cannot make a similar election. As a result, our consolidated financial statements may not be comparable to companies that comply with new or revised accounting pronouncements as of public company effective dates.
We will incur significant increased costs and management resources as a result of operating as a public company.
As a public company, we will incur significant legal, accounting, compliance and other expenses that GreenLight did not incur as a private company and that do not appear in our historical consolidated financial statements. These expenses may increase even more after we are no longer an “emerging growth company.” Our management and other personnel will need to devote a substantial amount of time and incur significant expense in connection with compliance initiatives. For example, we will need to implement additional internal controls, both generally and to address the material weaknesses discussed in “”, and disclosure controls and procedures. As a public company, we will bear all of the internal and external costs of preparing and distributing periodic public reports in compliance with our obligations under the securities laws.
Risks Relating to Our Business and Industry
In addition, regulations and standards relating to corporate governance and public disclosure, including the Sarbanes-Oxley Act, and the related rules and regulations implemented by the SEC and the Nasdaq Stock Market, LLC (“
Nasdaq
”), have increased legal and financial compliance costs and will make some compliance activities more time-consuming. For example, Nasdaq imposes requirements to obtain stockholder approval for the issuance of equity securities in a variety of circumstances, and this requirement can limit the financing alternatives available to us and thereby increase the cost of capital, which could reduce shareholder returns. We intend to invest resources to comply with evolving laws, regulations and standards, and this investment will result in increased general and administrative expenses and may divert management’s time and attention from our other business activities. If our efforts to comply with new laws, regulations and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to practice, regulatory authorities may initiate legal proceedings against us, and our business may be harmed. In the future, it may be more expensive or more difficult for us to obtain director and officer liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. These factors could also make it more difficult for us to attract and retain qualified members of our board of directors, particularly to serve on our audit committee and compensation committee, and qualified executive officers.96
Risks Related to the Business Combination and ENVI
The former GreenLight stockholders have significant influence over us.
Upon the completion of the Business Combination, the GreenLight stockholders collectively owned approximately 85% of the outstanding New GreenLight Common Stock. Some of these persons or entities may have interests different than yours. For example, because many of these stockholders purchased their shares at prices substantially below the agreed-upon valuation of the consideration issued in the Business Combination and have held their shares for a longer period, they may be more interested in selling New GreenLight to an acquirer than other investors or they may want us to pursue strategies that deviate from the interests of other stockholders.
Investors in New GreenLight will not have the same benefits as an investor in an underwritten public offering.
Upon the completion of the Business Combination, the directors, officers and stockholders of GreenLight, a private company, obtained control of New GreenLight, a public company, and the business of GreenLight became the business of New GreenLight. In this respect, the Business Combination was an indirect path for GreenLight to obtain the benefits of becoming a publicly listed company. However, the Business Combination was not an underwritten initial public offering of GreenLight’s securities and differed from an underwritten initial public offering in several significant ways, which include, but are not limited to, the following:
Like other business combinations and spin-offs, in connection with the Business Combination, investors did not receive the benefits of the diligence performed by the underwriters in an underwritten public offering. Investors in an underwritten public offering may benefit from the role of the underwriters in such an offering. In an underwritten public offering, an issuer initially sells its securities to the public market via one or more underwriters, who distribute or resell such securities to the public. Underwriters have liability under the U.S. securities laws for material misstatements or omissions in a registration statement pursuant to which an issuer sells securities. Because the underwriters have a “due diligence” defense to any such liability by, among other things, conducting a reasonable investigation, the underwriters and their counsel customarily conduct a due diligence investigation of the issuer. Due diligence entails engaging legal, financial and/or other professionals to investigate the accuracy of an issuer’s disclosure regarding, among other things, its business and financial results. In underwritten public offerings, investors have the benefit of such diligence in making their investment decision. The Business Combination did not involve any underwriters and, accordingly, no underwriter has ever conducted due diligence with respect to the business of GreenLight. Our investors must rely on the information disclosed in our filings with the Commission and will not have the benefit of any prior independent review and investigation of the type normally performed by an underwriter in a public securities offering.
While sponsors, private investors and management in a business combination undertake a certain level of due diligence, it is not necessarily the same level of due diligence undertaken by an underwriter in a public securities offering and, therefore, there is a heightened risk of an incorrect valuation of GreenLight’s business or material misstatements or omissions in this annual report.
In addition, because no underwriters were engaged in connection with the Business Combination, there was no traditional “roadshow” or book-building process before the closing of the Business Combination, and no underwriters set any initial public offering price to facilitate price discovery with respect to our securities after the closing of the Business Combination. Therefore, buy and sell orders for our securities, whether submitted before or after the closing of the Business Combination, have not had the benefit of being informed by a published price range or a price at which any underwriters initially sold shares to the public, as would be the case in an underwritten initial public offering. Moreover, as of the date of this annual report, there are no underwriters assuming risk in connection with an initial resale of our securities or helping to stabilize, maintain or affect the public price of any our securities, including those offered hereby. Moreover, we do not intend to engage in, and have not requested and do not intend to request, directly or indirectly, financial advisors to engage in, any special selling efforts or stabilization or price support activities in connection with any of our securities. In addition, securities analysts of major brokerage firms may not provide coverage of New GreenLight. No assurance can be
97
given that brokerage firms will, in the future, want to conduct any offerings on our behalf. All of these differences from an underwritten public offering of GreenLight’s securities could result in a more volatile price for our securities.
These differences from an underwritten public offering may present material risks to unaffiliated investors that would not exist if GreenLight had become a publicly listed company through an underwritten initial public offering instead of upon completion of the Business Combination.
Our future depends on the continued contributions of our senior management team and our ability to attract and retain other highly qualified personnel; in particular, Andrey Zarur, our President and Chief Executive Officer, is critical to our future vision and strategic direction.
Our success depends in large part on our ability to attract and retain high-quality management in sales, market access, product development, software engineering, marketing, operations, finance and support functions, especially in the Boston and Rochester areas. We compete for qualified technical personnel with other life sciences and biotechnology companies. Competition for qualified employees is intense in these industries, and the loss of even a few qualified employees, or an inability to attract, train, retain and motivate additional highly skilled employees required for the planned expansion of our business could harm our operating results and impair our ability to grow. The loss of one or more of our key employees, and any failure to have in place and execute an effective succession plan for key executives, could seriously harm our business.
As we continue to grow, we may be unable to continue to attract or retain the personnel needed to maintain our competitive position. To attract, train and retain key personnel, we use various measures, including competitive compensation and benefit packages (including an equity incentive program), which may require significant investment. These measures may not be enough to attract and retain the personnel required to operate our business effectively and efficiently.
Moreover, if the perceived value of our equity awards declines, it may materially and adversely affect our ability to attract and retain key employees. If we do not maintain the necessary personnel to accomplish our business objectives, we may experience staffing constraints that materially and adversely affect our ability to support programs and operations.
Many of our employees may receive proceeds from sales of our equity in the public markets, which may reduce their motivation to continue to work for us.
In addition, our future also depends on the continued contributions of our senior management team and other key personnel, each of whom would be difficult to replace. In particular, Dr. Andrey Zarur, our President and Chief Executive Officer, is critical to our future vision and strategic direction. We rely on our executive team in the areas of operations, research and development, commercial, and general and administrative functions. We also do not maintain key person life insurance for our key employees.
From time to time, there may be changes in our senior management team that may be disruptive to our business. If our senior management team, including any new hires that we may make, fails to work together effectively and to execute our plans and strategies on a timely basis, our business, results of operations and financial condition could be harmed.
Investors should not rely on outdated financial projections.
In connection with the Business Combination, we disclosed certain projections of GreenLight’s potential financial performance in future years. As previously disclosed, these projections were prepared solely for GreenLight’s internal use, capital budgeting and other management purposes, were finalized as of June 30, 2021 and were not updated to reflect events after that date. Also as previously disclosed, the projections were not
98
prepared with a view toward public disclosure or with a view toward complying with U.S. GAAP, the published guidelines of the SEC or the guidelines established by the American Institute of Certified Public Accountants for preparation and presentation of prospective financial information. Readers were cautioned not to rely on the prospective financial information because actual results are likely to differ materially from the prospective financial information. In light of the passage of time since June 30, 2021, the projections have become outdated and do not necessarily represent the current views of management. We reiterate our prior caution not to rely on the previously published and now outdated financial projections. We have not undertaken any obligation to publish any financial projections.
The unaudited pro forma combined financial information that we have filed with the Commission may not be indicative of what our actual financial position or results of operations would have been.
The unaudited pro forma combined financial information that we have filed with the Commission was presented for illustrative purposes only and does not necessarily reflect what New GreenLight’s financial condition or results of operations would have been had the Business Combination and the PIPE Financing occurred on the dates indicated. Further, the unaudited pro forma combined financial information also may not be useful in predicting the future financial condition and results of operations of New GreenLight. The actual financial position and results of operations may differ significantly from the pro forma amounts reflected therein due to a variety of factors. The unaudited pro forma adjustments represent management’s estimates based on information available as of the date of the unaudited pro forma combined financial statements and are subject to change as additional information becomes available and analyses are performed.
We are subject to changing laws and regulations regarding regulatory matters, corporate governance and public disclosure that have increased and will continue to increase costs and the risk of noncompliance.
We are subject to rules and regulations by various governing bodies, including, for example, the SEC, which is charged with the protection of investors and the oversight of companies whose securities are publicly traded, and to new and evolving regulatory measures under applicable law. Our efforts to comply with new and changing laws and regulations have resulted, and will likely continue to result, in increased general and administrative expenses and a diversion of management time and attention from business operations.
Moreover, because these laws, regulations and standards are subject to varying interpretations, their application in practice may evolve over time as new guidance becomes available. This evolution may result in continuing uncertainty regarding compliance matters and additional costs necessitated by ongoing revisions to our disclosure and governance practices. If we fail to address and comply with these regulations and any subsequent changes, we may be subject to penalty and our business may be harmed.
During 2021, ENVI identified two material weaknesses in its internal control over financial reporting, which may result in material misstatements or restatements of our consolidated financial statements or cause us to fail to meet our periodic reporting obligations.
On April 12, 2021, the Acting Director of the Division of Corporation Finance and Acting Chief Accountant of the SEC issued a statement regarding the accounting and reporting considerations for warrants issued by special purpose acquisition companies entitled “” (the “”). In light of the SEC Staff Statement, ENVI’s management concluded that ENVI’s audited balance sheet as of January 19, 2021 (the “”) should be revised to present ENVI’s warrants as liabilities. In connection with the foregoing development, ENVI identified a material weakness in its internal control over financial reporting.
Staff Statement on Accounting and Reporting Considerations for Warrants Issued by Special Purpose Acquisition Companies
SEC Staff Statement
IPO Balance Sheet
Additionally, ENVI initially recorded a portion of the ENVI Class A Common Stock subject to possible redemption, which was issued in its IPO, in permanent equity. In accordance with SEC Staff guidance on redeemable equity instruments, ASC“”, and EITF Topic
480-10-S99,
Distinguishing Liabilities from Equity
99
D-98,
“Classification and Measurement of Redeemable Securities
re-evaluated
the effectiveness of its disclosure controls and procedures as of June 30, 2021. Based upon that evaluation, ENVI concluded that the misclassification of the ENVI Class A Common Stock was quantitatively material to individual line items within the balance sheet but was not material to its reported financial position and was qualitatively immaterial to ENVI’s financial statements. ENVI’s management further concluded that the misstatement was not indicative of a pervasive issue in ENVI’s internal control, had no impact on its statement of cash flows, did not impact any other balance sheet line items other than total stockholders’ equity and ENVI Class A Common Stock subject to redemption, and was not disclosed in any other Exchange Act filings other than ENVI’s IPO Balance Sheet and Forms10-Q
for the periods ended March 31, 2021 and June 30, 2021. Based upon the foregoing, and due to the industry-wide issues and related insufficient risk assessment of the underlying accounting for certain instruments, ENVI’s management concluded that the misclassification of the ENVI Class A Common Stock represented a significant deficiency. A significant deficiency is a deficiency, or a combination of deficiencies, in internal control over financial reporting that is less severe than a material weakness, yet important enough to merit attention by those responsible for oversight of a company’s financial reporting.As of September 30, 2021, in light of the prior reclassification of warrants from equity to liability, as well as the reclassification of redeemable Class A Common Stock as temporary equity, ENVI identified a second material weakness in its internal control over financial reporting relating to accounting for complex financial instruments.
Management of New GreenLight was unable to conclude that the foregoing material weaknesses had been remediated as of December 31, 2021 and therefore concluded that ENVI’s internal control over financial reporting was not effective as of that date.
Risks Related to Redemptions
If we file a bankruptcy petition or an involuntary bankruptcy petition is filed against us that is not dismissed, a bankruptcy court may seek to recover proceeds that we distributed to our public stockholders from our trust account, and we and our board may be exposed to claims of punitive damages.
If we file a bankruptcy petition or an involuntary bankruptcy petition is filed against us that is not dismissed, the proceeds that we distributed to our public stockholders from our trust account could be viewed under applicable debtor/creditor and/or bankruptcy laws as either a “preferential transfer” or a “fraudulent conveyance.” As a result, a bankruptcy court could seek to recover all amounts received by our stockholders. In addition, our board of directors may be viewed as having breached its fiduciary duty to our creditors and/or having acted in bad faith, thereby exposing the directors and us to claims of punitive damages, by paying public stockholders from the trust account prior to addressing the claims of creditors. We and our directors may not have sufficient resources to satisfy any such claims in full, or at all.
Risks Related to Being a Public Benefit Corporation
Although we are a public benefit corporation, we cannot provide any assurance that we will achieve our PBC Purpose.
In connection with the Business Combination, we became a public benefit corporation under Delaware law, and our public benefit purpose is set forth in the Charter. Our public benefit purpose is to improve the public health and wellbeing of people and the environment by engineering, developing and commercializing biological
100
products that can reduce chemicals in our environment and promote health through delivery of high quality, affordable products that improve outcomes for people and the planet. As a public benefit corporation, we are required to operate in a responsible and sustainable manner, balancing our stockholders’ pecuniary interests, the best interests of those materially affected by our conduct and our public benefit purpose. When we use the term ‘sustainable,’ we refer to our efforts to align economic development with environmental protection and human well-being as well as our obligations as a public benefit corporation under § 362(a) of the Delaware General Corporation Law. There is no assurance that we will be able to achieve our public benefit purpose or that the expected positive impact from being a public benefit corporation will be realized, which could have a material adverse effect on our reputation, which in turn may have a material adverse effect on our business, results of operations and financial condition.
As a public benefit corporation, our focus on a specific public benefit purpose and producing a positive effect for society may negatively impact our financial performance.
Unlike traditional corporations, whose directors have a fiduciary duty to focus exclusively on maximizing stockholder value, our directors have a fiduciary duty to balance (i) the pecuniary interests of our stockholders, (ii) the best interests of those materially affected by our conduct and (iii) our public benefit purpose, as set forth in the Charter. Therefore, we may take actions that we believe will be in the best interests of those stakeholders materially affected by our public benefit purpose even if those actions do not maximize our financial results. While we intend for this public benefit designation and obligation to provide an overall net benefit to us and our stakeholders, it could instead cause us to make decisions and take actions without seeking to maximize the income generated from our business, and hence available for distribution to our stockholders. Our pursuit of longer-term
or non-pecuniary benefits
may not materialize within the timeframe we expect or at all, yet may have an immediate negative effect on any amounts available for distribution to our stockholders. Accordingly, being a public benefit corporation could have a material adverse effect on our business, results of operations and financial condition, which in turn could cause our stock price to decline.Also, as a public benefit corporation, because the New GreenLight Board is required by the DGCL to manage or direct our business and affairs in a manner that balances the pecuniary interests of our stockholders, the best interests of those materially affected by our conduct, and our public benefit purpose, we believe that our public benefit corporation status could make it more difficult for another party to obtain control of us without maintaining our public benefit corporation status and purpose. While Delaware common law, as stated in, 506 A.2d 173 (Del. 1986), and related cases, may impose upon directors of a traditional corporation a duty to maximize short-term stockholder value in certain ‘sale of the company’ transactions, a public benefit corporation board’s decision-making would not be subject to those same constraints. Any of the foregoing provisions could limit the price that investors might be willing to pay in the future for shares of our capital stock, and they could deter potential acquirers of our company, thereby reducing the likelihood that you would receive a premium for your shares of New GreenLight Common Stock in an acquisition.
Revlon, Inc. v. MacAndrews
& Forbes Holdings, Inc.
Further, public benefit corporations may not be attractive targets for activists or hedge fund investors because new directors would still have to consider and give appropriate weight to the public benefit along with shareholder value, and shareholders committed to the public benefit can enforce this through derivative suits. By requiring that boards of directors of public benefit corporations consider constituencies in addition to shareholder value, Delaware public benefit corporation law could potentially make it easier for a board to reject a hostile bid, even where the takeover would provide the greatest short-term financial yield to investors.
Our directors have a fiduciary duty to consider not only our stockholders’ interests, but also our specific public benefit and the interests of other stakeholders affected by our actions. If a conflict between such interests arises, there is no guarantee such a conflict would be resolved in favor of our stockholders.
While directors of traditional corporations are required to make decisions they believe to be in the best interests of their stockholders, directors of a public benefit corporation have a fiduciary duty to consider not only
101
the stockholders’ interests, but also the company’s specific public benefit and the interests of other stakeholders affected by the company’s actions. Under Delaware law, directors are shielded from liability for breach of these obligations if they make informed and disinterested decisions that serve a rational purpose. Thus, unlike traditional corporations whose directors must focus exclusively on stockholder value, our directors are not merely permitted, but obligated, to consider our specific public benefit and the interests of other stakeholders. In the event of a conflict between the interests of our stockholders and the interests of our specific public benefit or our other stakeholders, our directors must only make informed and disinterested decisions that serve a rational purpose; thus, there is no guarantee such a conflict would be resolved in favor of our stockholders, which could have a material adverse effect on our business, results of operations and financial condition, which in turn could cause our stock price to decline.
As a public benefit corporation, we are required to comply with various new reporting requirements, which, even if complied with, could result in harm to our reputation.
As a public benefit corporation, we are required to publicly disclose a report at least biennially on our overall public benefit performance and on our assessment of our success in achieving our specific public benefit purpose. If we are not timely or are unable to provide this report, or if the report is not viewed favorably by parties doing business with us or regulators or others reviewing our credentials, our reputation and status as a public benefit corporation may be harmed and the value of our stock could decrease as a result.
While not required by Delaware law or the terms of the Charter, we may elect to have our environmental, social and governance (“
ESG
”) performance assessed against ESG standards, including proprietary criteria established byindependent non-profit organizations.
For example, we may seek a Certified B Corporation certification. The requirements for these certifications may change over time. These standards may not be appropriately tailored to the legal requirements of publicly traded companies or to the operational requirements of larger companies. Additionally, our management team might have to spend significant time considering and meeting such certifications or such standards (including preparation of relevant applications and reports) and therefore will be spending less time on operating our business. Further, our reputation could be harmed if we obtain and then lose ESG certifications, whether by our choice or by our failure to meet certification requirements, or if that change in status were to create a perception that we are more focused on financial performance and are no longer as committed to the values shared by certifying organizations. Likewise, our reputation could be harmed if scores given to us by certifying organizations decline, since this might create a perception that we have slipped in our satisfaction of such standards. Similarly, our reputation could be harmed if we take actions that are perceived to be misaligned with our values.As a public benefit corporation, we may be subject to increased derivative litigation concerning our duty to balance stockholder and public benefit interests, the occurrence of which may have an adverse impact on our financial condition and results of operations.
Stockholders of a Delaware public benefit corporation (if they, individually or collectively, own at least 2% of its outstanding capital stock or at least $2.0 million in market value) are entitled to file a derivative lawsuit claiming that its directors failed to balance stockholder and public benefit interests. This potential liability does not exist for traditional corporations. Therefore, we may be subject to the possibility of increased derivative litigation, which would require the attention of management and, as a result, may adversely impact management’s ability to effectively execute our strategy. Any such derivative litigation may be costly and have an adverse impact on our financial condition and results of operations.
Our status as a public benefit corporation could make an acquisition of our company, which may be beneficial to our stockholders, more difficult.
While Delaware common law, as stated in, and related cases, may impose upon directors of a traditional corporation a duty to maximize short-term stockholder value in
Revlon, Inc. v. MacAndrews
& Forbes Holdings, Inc.
102
certain ‘sale of the company’ transactions, a public benefit corporation board’s decision-making would not be subject to those same constraints. Our Board could reject a bid to acquire New GreenLight in favor of pursuing other stakeholder interests or the specified public benefit, to the detriment of stockholders. Consideration of these competing interests would not preclude our Board from accepting a bid that maximizes short-term stockholder value. Rather, our Board could weigh the merits of accepting the short-term value offered by a bid against other options that may generate greater long-term value or have other meaningful effects on those materially affected by our conduct or public benefit purpose and, if appropriate, could accept a bid that does not maximize short-term value.
ITEM 1B. Unresolved Staff Comments | ||
None.
ITEM 2. Properties | ||
Our principal facilities are located in the metropolitan area of Boston, Massachusetts, Rochester, New York, and Research Triangle Park, North Carolina. We lease all of our facilities.
Our corporate headquarters are located in Medford, Massachusetts, where we lease an aggregate of approximately 50,000 square feet of office and laboratory space. Our leases for this facility expire between February 2024 and February 2025.
Agricultural Manufacturing Facilities
Our manufacturing facilities for our dsRNA agricultural products are located in Rochester, New York. Our lease for this facility commenced in January 2020 and expires in March 2025. Our existing manufacturing operations occupy approximately 17,000 square feet and include two
2,000-liter
bioreactors, one of which became operational in the third quarter of 2021.We expect that our operational bioreactor will provide sufficient manufacturing capacity for our current projected near-term needs for our dsRNA agricultural products and that we would activate the second bioreactor when needed to address an increase in production demand. We estimate that we can currently produce 500 kg of dsRNA per year and that, if the second bioreactor were brought online, we could produce 1,000 kg of dsRNA per year. We estimate that the activation of the second bioreactor would take approximately six months and require an additional investment of approximately $0.6 million, none of which had been incurred as of December 31, 2021. We are currently designing an expansion of our Rochester facility to further increase our manufacturing capacity to up to 30,000 liters of dsRNA for agricultural use.
In the third quarter of 2021, we leased approximately 5,557 square feet of additional laboratory space in the Rochester facility for dsRNA process development. We estimate that this laboratory space will be available for use in the third quarter of 2022 and will require an aggregate investment of approximately $0.5 million, none of which had been incurred as of December 31, 2021.
Agricultural Laboratory and Greenhouse
We currently lease approximately 14,000 square feet of laboratory, office and greenhouse space for our agricultural operations at our facility in Research Triangle, North Carolina. Our lease for this facility commenced in January 2019 and expires in December 2026.
On September 30, 2021, we entered into a new lease for approximately 63,000 square feet of laboratory, office and greenhouse space on the same campus as our existing Research Triangle Park facility. The lease expires 11 years after an occupancy date determined in accordance with the terms of the lease. We expect to
103
relocate our current operations in Research Triangle Park to this new facility in the second half of 2022. We estimate that the total cost to build out the new facility will be approximately $7.0 million, none of which had been incurred as of December 31, 2021.
Human Health Facilities
In addition to research and development conducted at our headquarters, we conduct a portion of our human health research and development activities in approximately 19,000 square feet of laboratory and office space in Woburn, Massachusetts. Our lease for this facility commenced in November 2020 and expires in February 2024.
We recently leased approximately 1,500 square feet in Burlington, Massachusetts for clean room facilities for
pre-clinical
and early-phase clinical material manufacturing for our human health program. Our lease for this facility commenced in August 2021 and expires in August 2023. We expect that these facilities will allow us to manufacture mRNA clinical materials at three-liter scale. We anticipate that the facilities will begin production by the end of 2021. We expect that the total cost to build out this facility, including GMP readiness, will be approximately $2.5 million, of which $1.9 million had been incurred as of December 31, 2021.We intend to produce mRNA clinical materials for early-stage clinical trials at our Burlington facility, and we anticipate that this facility will provide sufficient manufacturing capacity for our current projected near-term needs.
We currently anticipate that we will contract with third parties to produce mNRA clinical materials for amounts needed in excess of the capacity of our Burlington facility, including production of our”..
COVID-19
vaccine candidate. In November 2021, we entered into agreements with Samsung Biologics Co., Ltd. to provide manufacturing services to fulfill our mRNA production needs. For more information regarding our agreements with Samsung Biologics Co., Ltd., see “ —Our Manufacturing Platform — Our manufacturing platform for human health (mRNA)
We believe our facilities are adequate and suitable for our current needs. To support future organic growth oractivity, we may enter into new leases, assume lease obligations, or acquire property both domestically and internationally. We believe that suitable or alternative space will be available if and when needed.
merger-and-acquisition
ITEM 3. Legal Proceedings | ||
From time to time, we may become involved in legal proceedings arising in the ordinary course of our business. The outcome of legal proceedings cannot be predicted with certainty, and the resolution of these matters could materially affect our business, financial condition or results of operations. We are not currently a party to any legal proceedings the outcome of which, if determined adversely to us, would individually or in the aggregate reasonably be expected to have a material adverse effect on our business, financial condition or results of operations.
ITEM 4. Mine Safety Disclosures | ||
Not applicable.
104
PART II
ITEM 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | ||
Market Information
The Common Stock and the Public Warrants are listed on Nasdaq under the symbols “GRNA” and “GRNAW,” respectively. Prior to the consummation of the Business Combination, the ENVI Class A Common Stock, ENVI Units and ENVI Public Warrants were traded on Nasdaq under the symbols “ENVI,” “ENVIU” and “ENVIW,” respectively.
Holders
As of March 15, 2022, there were 152 holders of record of our Common Stock. The number of holders of record does not include “street name” holders or beneficial holders whose shares of Common Stock are held of record by banks, brokers and other financial institutions.
Dividend Policy
We have not paid any cash dividends to date. We currently intend to retain all available funds and future earnings, if any, for the operation and expansion of our business and do not anticipate declaring or paying any dividends in the foreseeable future. The payment of cash dividends in the future will depend on our revenues and earnings, if any, capital requirements and general financial condition. The payment of any cash dividends will be within the discretion of our Board at such time. The terms of our existing loan agreements generally preclude us from paying cash dividends without consent. Our ability to declare dividends may also be limited by restrictive covenants under any future debt financing agreements.
Use of Proceeds from the IPO
ENVI’s registration statement on Form”). The underwriting fees and expenses were paid to ENVI’s qualified independent underwriter, selling group members and other third-party service providers unaffiliated with ENVI. For more information regarding the IPO and the trust account, see the Explanatory Note at the beginning of this Annual Report.
S-1
(SEC FileNo. 333-251593)
for its IPO was declared effective on January 13, 2021. Of total proceeds of $207.0 million, $250,000 was used to pay underwriting fees and expenses, and the remaining net proceeds of $206,750,000 were placed in the trust account (the “IPO Net Proceeds
In connection with the closing of the Business Combination, 19,489,626 shares of ENVI Class A Common Stock were redeemed for an aggregate payment of approximately $194.9 million. The redemption price was paid from the IPO Net Proceeds held in the trust account. The remaining IPO Net Proceeds of approximately $12.1 million were disbursed to New GreenLight, which used those funds to pay a portion of the expenses incurred in connection with the Business Combination.
The expenses of the Business Combination included payments of approximately $5.8 million to Canaccord Genuity LLC for services rendered in connection with the Business Combination pursuant to the terms of the Business Combination Marketing Agreement. Canaccord is an affiliate of the Sponsor and the employer of Daniel Coyne, ENVI’s President and Chief Executive Officer and a director of ENVI until the Closing Date, Marc Marano, ENVI’s Chief Financial Officer and Treasurer until the Closing Date, and Jennifer E. Pardi, a director of ENVI and a current director of New GreenLight. The balance of the IPO Net Proceeds was used to pay a portion of the fees and expenses of SVB Leerink LLC and Credit Suisse Securities (USA) LLC for their services as placement agents for the PIPE Financing.
105
ITEM 6. | Removed and reserved. |
ITEM 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
The following discussion and analysis of ENVI’s financial condition and results of operations should be read in conjunction with ENVI’s audited consolidated financial statements and the notes related thereto, which are presented at the end of this Annual Report beginning on page
F-1.
This discussion and analysis includes forward-looking statements, and actual results may differ materially from those anticipated in these forward-looking statements as a result of many factors, including those set forth under “Cautionary Note Regarding Forward-Looking Statements,” “Item 1A. Risk Factors” and elsewhere in this Annual Report.Overview
ENVI was incorporated on July 2, 2020 as a special purpose acquisition company, a type of blank check company formed for the purpose of effecting a merger, capital stock exchange, asset acquisition, stock purchase, reorganization, or other similar business combination with one or more target businesses. ENVI was sponsored by CG Investments Inc. VI (the “”).
Sponsor
Until the consummation of the Business Combination on February 2, 2022, ENVI’s activities consisted principally of organizational activities, an initial public offering and related private placement, a search for one or more businesses to acquire, and the negotiation, execution, performance and consummation of the Business Combination, including the PIPE Financing.
Initial Public Offering and Private Placement
On January 19, 2021, ENVI consummated its IPO of 20,700,000 Units, including 2,700,000 Units issued to the underwriters upon full exercise of their over-allotment option. Each Unit consisted of one share of ENVI Class A Common Stock and”) at a purchase price of $1.00 per Private Placement Warrant, generating gross proceeds of $2.0 million. At the closing of the IPO, ENVI also issued 600,000 Private Placement Warrants to the Sponsor and 50,000 Private Placement Warrants to each of its three independent directors (together with HB Strategies and the Sponsor, the “”). A total of $207.0 million, comprised of $206,750,000 of the proceeds from the IPO and $250,000 of the proceeds of the sale of the Private Placement Warrants, was placed in the trust account maintained by Continental Stock Transfer & Trust Company, acting as trustee.
one-half
of one warrant, with each whole warrant entitling the holder thereof to purchase one share of Class A Common Stock for $11.50 per share. The Units were sold at a price of $10.00 per Unit, generating gross proceeds to ENVI of $207.0 million. Simultaneously with the closing of the IPO, ENVI completed the private sale of an aggregate of 2,000,000 Private Placement Warrants to HB Strategies, LLC (“HB Strategies
Initial Stockholders
Business Combination
On August 9, 2021, ENVI entered into the Business Combination Agreement with Merger Sub and GreenLight, under which ENVI agreed to acquire GreenLight in exchange for aggregate consideration of up to 120,000,000 shares of Class A Common Stock. On February 2, 2022, pursuant to the terms of the Business Combination Agreement, Merger Sub merged with and into GreenLight, with GreenLight surviving the merger as a wholly owned subsidiary of ENVI. In connection with the consummation of the Merger on the Closing Date, ENVI changed its name to GreenLight Biosciences Holdings, PBC and became a public benefit corporation.
In accordance with the terms and subject to the conditions of the Business Combination Agreement, at the effective time of the Merger, each outstanding share of capital stock of GreenLight (other than treasury shares) was exchanged for shares of New GreenLight Common Stock, and outstanding GreenLight options and warrants to purchase shares of capital stock of GreenLight (whether vested or unvested) were converted into comparable options and warrants to purchase shares of New GreenLight Common Stock, in each case, based on an implied GreenLight fully diluted equity value of $1.2 billion. In connection with the consummation of the Business Combination, all of the issued and outstanding shares of ENVI Class A Common Stock and all of the issued and outstanding shares of ENVI Class B Common Stock became shares of New GreenLight Common Stock.
106
In the Business Combination, New GreenLight issued an aggregate of approximately 104.0 million shares of New GreenLight Common Stock, Rollover Options to purchase an aggregate of approximately 18.0 million shares of New GreenLight Common Stock and Assumed Warrants to purchase less than 0.1 million shares of New GreenLight Common Stock.
Consummation of PIPE Financing
In connection with the Business Combination, New GreenLight completed the sale and issuance of 12,425,000 shares of New GreenLight Common Stock in a private placement at a purchase price of $10.00 per share pursuant to the Subscription Agreements, which had been entered into between ENVI and the PIPE Investors either concurrently with the execution of the Business Combination Agreement or subsequently in November 2021.
Redemption of ENVI Class A Common Stock
Also in connection with the Business Combination, 19,489,626 shares of ENVI Class A Common Stock were redeemed for an aggregate payment of approximately $194.9 million. The redemption price was paid from the trust account, the remaining balance of the trust account was disbursed to New GreenLight, and the trust account was closed.
Proceeds of the Business Combination and PIPE Financing
New GreenLight’s gross proceeds from the Business Combination and the PIPE Financing totaled approximately $136.4 million, which included approximately $12.1 million of funds held in the trust account (after giving effect to redemptions) and approximately $124.3 million of proceeds from the PIPE Financing, inclusive of $35.25 million previously advanced to GreenLight, as described in more detail in “” as of and for the years ended December 31, 2021 and 2020 (the “”), which is filed as Exhibit 99.2 to New GreenLight’s Current Report on Form
Management’s Discussion and Analysis of Financial Condition and Results of Operations of GreenLight
GreenLight MD&A
8-K
filed on the date hereof and is incorporated herein by reference. The gross proceeds do not reflect estimated aggregate transaction expenses and other costs related to the Business Combination, the PIPE Financing and other transactions of approximately $25.0 million.Warrant Forfeiture
Concurrently with the execution of the Business Combination Agreement, the Initial Stockholders and GreenLight entered into an agreement (the “”), pursuant to which HB Strategies and the Sponsor agreed that, if more than 25% of the shares of ENVI Class A Common Stock were redeemed pursuant to ENVI’s Amended and Restated Certificate of Incorporation in effect prior to the Business Combination (the “”), then they would forfeit 25% of the warrants issued to the Initial Stockholders. At the closing of the Business Combination, pursuant to the terms of the Sponsor Letter Agreement, HB Strategies and the Sponsor forfeited an aggregate of 687,500 Private Placement Warrants.
Sponsor Letter Agreement
Existing Charter
Results of Operations
From its inception until the consummation of the Business Combination, ENVI neither engaged in any operations nor generated any operating revenues. During such period, ENVI’s only activities were organizational activities, activities in connection with the IPO and related private placement, identifying a target company for a business combination, and activities in connection with the acquisition of GreenLight. ENVI generated
non-operating
income in the form of interest income on marketable securities held in the trust account. ENVI incurred expenses as a result of being a public company (for legal, financial reporting, accounting and auditing compliance), as well as for due diligence expenses.107
For the year ended December 31, 2021, ENVI had a net loss of $15.1 million, which consisted of general and administrative expenses of $13.1 million, a loss on initial issuance of the Private Placement Warrants of $1.3 million and a change in fair value of warrant liabilities of $0.7 million. Marketable securities held in the trust account earned interest of $12,391.
For the period from July 2, 2020 (inception) through December 31, 2020, ENVI had a net loss of $2,528, which consisted of formation and operating expenses.
Liquidity and Capital Resources; Going Concern
From its inception until the consummation of the Business Combination, ENVI’s principal sources of liquidity consisted of loans from affiliates and the proceeds of the issuance of the Private Placement Warrants. Although ENVI raised gross proceeds of $207.0 million in its IPO on January 19, 2021, substantially all of those proceeds were placed in the trust account and were not available for use in funding operations. Interest income prior to the consummation of the Business Combination was not material. ENVI had no other capital resources or sources of liquidity.
On September 4, 2020, HB Strategies agreed to loan ENVI up to $0.3 million pursuant to an unsecured promissory note. The note was
non-interest
bearing and was payable on the earlier of March 31, 2021 or the consummation of the IPO. From September 4, 2020 to the closing of the IPO, ENVI borrowed $0.3 million to fund operating expenses. ENVI repaid the full amount of the loan at the closing of the IPO on January 19, 2021.As described above under “,” on January 19, 2021, ENVI received gross proceeds of $2.0 million from the issuance of the Private Placement Warrants simultaneously with the closing of the IPO. The proceeds of this financing were used to fund operating expenses and the repayment of the $0.3 million loan from HB Strategies.
— Overview — Initial Public Offering and Private Placement
On August 9, 2021, HB Strategies loaned ENVI $0.5 million pursuant to an unsecured promissory note. The note was
non-interest
bearing and was due and payable on the earlier of January 19, 2022 or the closing of the Business Combination. The proceeds of the loan were used to fund operating expenses. In connection with the consummation of the Business Combination, HB Strategies forgave the full amount of the loan.Cash Flows from Operating Activities
For the year ended December 31, 2021, cash used in operating activities was $1.8 million. Net loss of $15.1 million included a
non-cash
loss on initial issuance of Private Placement Warrants of $1.3 million,non-cash
charges related to the change in fair value of the warrant liabilities of $0.7 million and transaction costs associated with the warrants of $0.1 million. Net changes in operating assets and liabilities, consisting primarily of an increase in accounts payable and accrued expenses, provided $11.2 million of cash for operating activities. No cash was used in operating activities for the year ended December 31, 2020.Cash Flows from Investing Activities
For the year ended December 31, 2021, net cash used in investing activities was $207.0 million, which consisted of investments in the trust account. No cash was used in investing activities for the year ended December 31, 2020.
Cash Flows from Financing Activities
For the year ended December 31, 2021, net cash provided by financing activities was $208.7 million, consisting primarily of net proceeds from the IPO of $206.8 million, proceeds from the issuance of the Private Placement Warrants of $2.0 million and proceeds from a loan from HB Strategies of $0.5 million, partially offset by
108
repayment of a loan from HB Strategies of $0.3 million and payment of offering costs of $0.2 million. For the year ended December 31, 2020, net cash provided by financing activities was $0.2 million, consisting primarily of proceeds from a loan from HB Strategies of $0.2 million, partially offset by payment of offering costs.
Cash Position
As of December 31, 2021, ENVI had cash of less than $0.1 million and marketable securities held in the trust account of $207.0 million, including an immaterial amount earned from interest income. At December 31, 2021, ENVI expected to use the amounts in the trust account to fund an uncertain amount of potential redemptions of ENVI Class A Common Stock in connection with the then-anticipated consummation of the Business Combination and to use any amounts remaining after such redemptions to satisfy its existing obligations, including $11.7 million of accounts payable and accrued expenses and $0.5 million of loans as of December 31, 2021, and to fund the future operations of GreenLight following the then-anticipated consummation of the Business Combination.
Upon consummation of the Business Combination on February 2, 2022, ENVI used the amounts in the trust account to redeem 19,489,626 shares of ENVI Class A Common Stock for approximately $194.9 million and to satisfy a portion of its existing obligations.
Accounting Predecessor; Known Trends and Uncertainties
As a result of the Business Combination, GreenLight is considered the accounting predecessor of New GreenLight. The audited consolidated financial statements of GreenLight as of and for the years ended December 31, 2021 and 2020 (the “”) have been filed as Exhibit 99.1 to the Current Report on Form
GreenLight Financial Statements
8-K
filed by New GreenLight on the date hereof and are incorporated herein by reference. For filings made after the Closing Date (other than this Annual Report), the consolidated financial statements of GreenLight will be the consolidated financial statements of New GreenLight.In light of the consummation of the Business Combination, the historical consolidated financial statements of ENVI ceased to be representative of the consolidated financial position, results of operations, stockholders’ equity and cash flows of New GreenLight. For information regarding the consolidated financial position, results of operations, stockholders’ equity and cash flows of New GreenLight as of and for the years ended December 31, 2021 and 2020, see the GreenLight Financial Statements. For management’s discussion and analysis of the GreenLight Financial Statements, including known trends and uncertainties regarding the future consolidated financial position, results of operations, stockholders’ equity and cash flows of New GreenLight, see the GreenLight MD&A, which is incorporated herein by reference.
Going Concern
As a result of the factors discussed above and in the GreenLight MD&A, including the subsection entitled “—,” in connection with New GreenLight’s assessment of going concern considerations in accordance with the Financial Accounting Standard Board’s (“”) Accounting Standards Update (“”),” New GreenLight management has determined that New GreenLight’s liquidity condition raises substantial doubt about its ability to continue as a going concern through one year after the date that ENVI’s consolidated financial statements are issued.
—Liquidity and Capital Resources
Funding Future
Operations;
Going Concern
FASB
ASU
2014-15,
“Disclosures of Uncertainties about an Entity’s Ability to Continue as a Going Concern
Off-Balance
Sheet Financing ArrangementsAs of December 31, 2021, ENVI had no obligations, assets or liabilities that would be considered
off-balance
sheet financing arrangements. Through December 31, 2021, ENVI did not participate in transactions109
that created relationships with unconsolidated entities or financial partnerships, often referred to as variable interest entities, for the purpose of facilitating
off-balance
sheet financing arrangements. Through December 31, 2021, ENVI had not entered into anyoff-balance
sheet financing arrangements, established any special purpose entities, guaranteed any debt or commitments of other entities, or purchased anynon-financial
assets.Contractual Obligations
As of December 31, 2021, ENVI did not have any long-term debt, capital lease obligations, operating lease obligations or long-term liabilities, other than warrant liabilities.
Pursuant to the Business Combination Marketing Agreement that ENVI entered into with Canaccord in connection with its IPO, ENVI engaged Canaccord as advisors in connection with a business combination to assist ENVI in arranging meetings with its stockholders to discuss the potential business combination and the target business’s attributes, introduce ENVI to potential investors that may be interested in purchasing its securities, assist ENVI in obtaining stockholder approval for the business combination and assist ENVI with the preparation of its press releases and public filings in connection with the business combination. Pursuant to the Business Combination Marketing Agreement, ENVI agreed to pay Canaccord for such services, upon the consummation of a business combination, a cash fee in an amount equal to 3.76% of the gross proceeds of the IPO. Pursuant to the terms of the Business Combination Marketing Agreement, no fee would have been due if ENVI had not completed a business combination. At the Closing of the Business Combination, the parties agreed to reduce the cash fee to $5.8 million, and such amount was paid.
Critical Accounting Policies
The preparation of financial statements and related disclosures in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, disclosure of contingent assets and liabilities at the date of the financial statements, and income and expenses during the periods reported. Actual results could differ materially from those estimates. New GreenLight has identified the following critical accounting policies with respect to the consolidated financial statements of ENVI included in this Annual Report.
Warrant Liabilities
ENVI accounted for the Warrants in accordance with the guidance contained in Accounting Standards Codification (“”)and 7F, under which the Warrants did not meet the criteria for equity treatment and were therefore recorded as liabilities. Accordingly, ENVI classified the Warrants as liabilities at their fair value and must adjust the Warrants to fair value at each subsequent balance sheet date. These liabilities are subject to
ASC
815-40-15-7D
re-measurement
at each balance sheet date until the Warrants are exercised, and any change in fair value will be recognized in ENVI’s statements of operations. The Private Placement Warrants, for which no observable trading price was available during the reported periods, were valued using a Modified Black-Scholes Option Pricing Model. The Public Warrants, for periods where no observable trading price was available, were valued using a Monte Carlo simulation. For periods subsequent to the detachment of the Public Warrants from the Units, the Public Warrant quoted market price was used as the fair value as of each relevant date.ENVI Class A Common Stock Subject to Possible Redemption
ENVI accounted for the ENVI Class A Common Stock subject to possible redemption in accordance with the guidance in ASC Topic 480, “.” Under this guidance, shares of common stock subject to mandatory redemption are classified as liability instruments and are measured at fair value. Conditionally redeemable common stock (including common stock that features redemption rights that are within the control of the holder or that is subject to redemption upon the occurrence of uncertain events not solely within the issuer’s control) is classified as temporary equity. At all other times, common stock is classified
Distinguishing Liabilities from Equity
110
as stockholders’ equity. The ENVI Class A Common Stock featured certain redemption rights that were considered to be outside of ENVI’s control and subject to the occurrence of uncertain future events. Accordingly, the ENVI Class A Common Stock subject to possible redemption was presented as temporary equity, outside of the stockholders’ (deficit) equity section of ENVI’s balance sheets. ENVI (and, after the Closing Date, New GreenLight) must recognize changes in redemption value immediately as they occur and adjust the carrying value of the ENVI Class A Common Stock subject to possible redemption at the end of each reporting period to equal the redemption value at the end of each reporting period. This method would view the end of the reporting period as if it were also the redemption date for the security.
Net Income (Loss) Per Common Share
At December 31, 2021, ENVI had two classes of shares, the ENVI Class A Common Stock and the ENVI Class B Common Stock. Income and losses are shared pro rata between the two classes of shares. Net income (loss) per share of common stock is calculated by dividing the net income (loss) by the weighted average shares of common stock outstanding for the respective period. Accretion associated with the redeemable shares of ENVI Class A Common Stock is excluded from income (loss) per common share, as the redemption value approximates fair value.
Recent Accounting Standards
In August 2020, the FASB issued ASU” (“”), to simplify accounting for certain financial instruments. ASU
2020-06,
“Contracts in Entity’s Own Equity (Subtopic
815-40)
ASU
2020-06
2020-06
eliminates the current models that require separation of beneficial conversion and cash conversion features from convertible instruments and simplifies the derivative scope exception guidance pertaining to equity classification of contracts in an entity’s own equity. The new standard also introduces additional disclosures for convertible debt and freestanding instruments that are indexed to and settled in an entity’s own equity. ASU2020-06
amends the diluted earnings per share guidance, including the requirement to use theif-converted
method for all convertible instruments. ASU2020-06
is effective January 1, 2022 and should be applied on a full or modified retrospective basis, with early adoption permitted beginning on January 1, 2021. New GreenLight is currently assessing the impact, if any, that ASU2020-06
would have on its financial position, results of operations or cash flows.Management of New GreenLight does not believe that any recently issued, but not yet effective, accounting standards, if currently adopted, would have a material effect on New GreenLight’s consolidated financial statements.
ITEM 7A. | Quantitative and Qualitative Disclosure About Market Risk |
Not applicable.
ITEM 8. | Financial Statements and Supplementary Data |
The financial statements required by this Item 8 are presented at the end of this Annual Report starting on page
F-1.
ITEM 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
Change in Independent Registered Public Accounting Firm
WithumSmith+Brown, PC (“
Withum
”) served as the independent registered public accounting firm of ENVI prior to the completion of the Business Combination. Accordingly, Withum was informed that the New GreenLight Board approved Withum’s dismissal as New GreenLight’s independent registered public accounting firm once it completes the audit of ENVI’s financial statements for the year ended December 31, 2021.111
On February 7, 2022, the audit committee (the “
Audit Committee
”) of the New GreenLight Board approved the engagement of Deloitte & Touche LLP (“Deloitte
”) as New GreenLight’s principal independent registered public accounting firm to audit the consolidated financial statements of New GreenLight for the year ended December 31, 2021.Neither the report of Withum on ENVI’s balance sheet as of January 19, 2021 nor the report of Withum on ENVI’s balance sheet as of December 31, 2020, the statements of operations, changes in stockholders’ equity and cash flows for the period from July 2, 2020 (inception) to December 31, 2020, and the related notes to the financial statements, contained an adverse opinion or a disclaimer of opinion or was qualified or modified as to uncertainty, audit scope or accounting principles, other than the restatement of ENVI’s balance sheet and the emphasis of matter regarding ENVI’s ability to continue as a going concern as of January 19, 2021.
During the period from July 2, 2020 (inception) to December 31, 2021, and the subsequent interim period through February 6, 2022, there were no “disagreements” (as such term is defined in Item 304(a)(1)(iv) of
Regulation S-K under
the Exchange Act) between ENVI and Withum on any matter of accounting principles or practices, financial statement disclosure or auditing scope or procedure, which disagreements, if not resolved to the satisfaction of Withum, would have caused it to make reference to the subject matter of the disagreements in its reports on ENVI’s financial statements.During the period from July 2, 2020 (inception) to December 31, 2021, and the subsequent interim period through February 6, 2022, there were no “reportable events” (as defined in Item 304(a)(1)(v) of
Regulation S-K under
the Exchange Act), other than the occurrence of material weaknesses in internal control over financial reporting for the quarterly periods ended March 31, 2021, June 30, 2021 and September 30, 2021 as a result of ENVI’s disclosure controls not being effective for such quarterly periods.Disclosures Regarding the New Independent Auditor
As described above, on February 7, 2022, the Audit Committee approved the engagement of Deloitte as New GreenLight’s principal independent registered public accounting firm. Deloitte served as the independent registered public accounting firm of GreenLight prior to the Business Combination. During the period from July 2, 2020 (inception) to December 31, 2021, and the subsequent interim period through February 6, 2022, New GreenLight did not consult with Deloitte with respect to (i) the application of accounting principles to a specified transaction, either completed or proposed, or the type of audit opinion that might be rendered on New GreenLight’s financial statements, and neither a written report nor oral advice was provided to New GreenLight that Deloitte concluded was an important factor considered by New GreenLight in reaching a decision as to any accounting, auditing or financial reporting issue, or (ii) any other matter that was the subject of a disagreement or a reportable event (as defined above).
ITEM 9A. | Controls and Procedures |
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures, as defined in Rules
13a-15(e)
and15d-15(e)
under the Securities Exchange Act of 1934, as amended, or the Exchange Act, which are designed to ensure that information required to be disclosed in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate to allow timely decisions regarding required disclosure.Under the supervision and with the participation of our Chief Executive Officer and Chief Financial Officer, our management has evaluated the effectiveness of ENVI’s disclosure controls and procedures as of
112
December 31, 2021, the end of the period covered by this annual report. Based on that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that ENVI’s disclosure controls and procedures were not effective as of December 31, 2021 as a result of the material weaknesses in its internal control over financial reporting which are described in more detail below.
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Our internal control over financial reporting is the process designed by and under the supervision of our Chief Executive Officer and Chief Financial Officer to provide reasonable assurance regarding the reliability of our financial reporting and the preparation of our consolidated financial statements for external reporting in accordance with accounting principles generally accepted in the United States of America. Management has evaluated the effectiveness of ENVI’s internal control over financial reporting using the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control-Integrated Framework (2013). Under the supervision and with the participation of our Chief Executive Officer and Chief Financial Officer, our management has assessed the effectiveness of ENVI’s internal control over financial reporting as of December 31, 2021 and concluded that it was not effective as a result of two material weaknesses in its internal control over financial reporting relating to the accounting for complex financial instruments. Specifically, ENVI initially failed to accord liability treatment to the public warrants issued in its IPO and, as a result, failed to record them as a liability in its audited balance sheet as of January 19, 2021. ENVI restated this audited balance sheet on January 7, 2022. Further, ENVI misclassified a quantitatively material portion of the redeemable ENVI Class A Common Stock as permanent rather than temporary equity in its balance sheets as of January 13, 2021, March 31, 2021 and June 30, 2021. ENVI restated these balance sheets, as well as other portions of its financial statements for the periods ended on the dates of such balance sheets, on November 24, 2021. As of September 30, 2021, in light of the reclassification of warrants from equity to liability and the reclassification of redeemable ENVI Class A Common Stock from permanent to temporary equity, ENVI concluded that it had a second material weakness in its internal control over financial reporting relating to accounting for complex financial instruments. For more information, see “”
Item 1A. Risk Factors — Risks Related to the Business Combination and ENVI —
During 2021, ENVI identified two material weaknesses in its internal control over financial reporting, which may result in material misstatements or restatements of our consolidated financial statements or cause us to fail to meet our periodic reporting obligations.
As a result of these material weaknesses, we performed additional analysis to determine that ENVI’s financial statements were prepared in accordance with GAAP. Accordingly, management believes that the financial statements included in this annual report present fairly in all material respects ENVI’s financial position, results of operations and cash flows for the period presented in accordance with GAAP.
Management has implemented remediation steps to improve our internal control over financial reporting. Specifically, we expanded and improved our review process for complex securities and related accounting standards. We plan to further improve this process by enhancing access to accounting literature, identification of third-party professionals with whom to consult regarding complex accounting applications, and consideration of additional staff with the requisite experience and training to supplement existing accounting professionals. However, management was unable to conclude that the foregoing material weaknesses had been remediated as of December 31, 2021.
This annual report does not include an attestation report of our independent registered public accounting firm regarding management’s assessment of internal control over financial reporting. Because we are an emerging growth company, no attestation report is required.
Changes in Internal Control Over Financial Reporting
Under the supervision and with the participation of our Chief Executive Officer and Chief Financial Officer, our management has evaluated changes in ENVI’s internal control over financial reporting that occurred during the period
113
covered by this annual report. Based on that evaluation, our Chief Executive Officer and Chief Financial Officer did not identify any change in ENVI’s internal control over financial reporting that has materially affected, or is reasonably likely to materially affect, ENVI’s internal control over financial reporting, except as described above.
Important Considerations
The effectiveness of our disclosure controls and procedures and our internal control over financial reporting is subject to various inherent limitations, including cost limitations, judgments used in decision making, assumptions about the likelihood of future events, the soundness of our systems, the possibility of human error, and the risk of fraud. Moreover, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions and the risk that the degree of compliance with policies or procedures may deteriorate over time. For example, the disclosure controls and procedures and internal control over financial reporting used by ENVI at December 31, 2021 were designed for a special purpose acquisition company and likely would be inadequate for an operating biotechnology company. Because of these limitations, there can be no assurance that any system of disclosure controls and procedures or internal control over financial reporting will be successful in preventing all errors or fraud or in making all material information known in a timely manner to the appropriate levels of management.
GreenLight Material Weaknesses
In connection with the preparation and audit of GreenLight’s consolidated financial statements as of December 31, 2021, three material weaknesses were identified in its internal control over financial reporting. For more information, see “”
Item 1A. Risk Factors — Risks Relating to Our Business and Industry — Our accounting predecessor, GreenLight, has identified material weaknesses in its internal controls of financial reporting. If we are unable to remediate the material weaknesses, or if we identify additional material weaknesses or otherwise fail to maintain effective internal control over financial reporting, this may result in material misstatements or restatements of our consolidated financial statements or cause us to fail to meet our periodic reporting obligations.
Had management conducted an evaluation of the effectiveness of GreenLight’s disclosure controls and procedures and internal control over financial reporting as of December 31, 2021, these material weaknesses would have prevented management from concluding that such disclosure controls and procedures and internal control over financial reporting were effective.
ITEM 9B. | Other Information |
None.
ITEM 9C. | Disclosure Regarding Foreign Jurisdictions that Prevent Inspections |
Not applicable.
114
PART III
ITEM 10. Directors, Executive Officers and Corporate Governance
The following table provides, as of the date of this annual report, certain information regarding the executive officers and directors of New GreenLight.
Name | Age | Position | ||||
Executive Officers | ||||||
| Andrey J. Zarur, Ph.D. | 51 | Chief Executive Officer, President and Class III Director | ||||
| Carole Cobb, M.B.A. | 64 | Chief Operating Officer | ||||
| Charu Manocha, M.B.A. | 55 | Chief People Officer | ||||
| Marta Ortega-Valle, M.B.A. | 49 | Chief Business Officer, Human Health | ||||
| Susan Keefe, M.B.A | 49 | Chief Financial Officer | ||||
| David Kennedy | 60 | General Counsel | ||||
| Amin Khan, Ph.D. | 59 | Chief Scientific Officer | ||||
| Mark Singleton, Ph.D. | 54 | Senior Vice President of Technology | ||||
Non-Employee Directors | ||||||
| Charles Cooney (1) | 77 | Class III Director | ||||
| Ganesh Kishore (2)(3) | 69 | Class III Director | ||||
| Eric O’Brien (2) | 49 | Class I Director | ||||
| Jennifer E. Pardi (1)(3) | 41 | Class I Director | ||||
| Martha Schlicher (1)(2) | 62 | Class II Director | ||||
| Matthew Walker (2)(3) | 40 | Class II Director | ||||
| (1) | Member of the audit committee. |
| (2) | Member of the compensation committee. |
| (3) | Member of the nominating and corporate governance committee. |
Executive Officers
Andrey Zarur, Ph.D.
a co-founder of
GreenLight, has served as the Chief Executive Officer and President and as a member of the Board of each of New GreenLight and GreenLight since February 2022 and August 2008, respectively. Dr. Zarur is alsothe co-founder of
Lumicell Inc., an oncology company delivering advanced imaging solutions for cancer surgery, and has served as its Chairman of the Board since January 2010.Dr. Zarur co-founded and
served as the Chairman of the Board of Solid Biosciences, Inc. (NASDAQ: SLDB), a gene therapy company targeting Duchenne muscular dystrophy, from February 2014 to June 2020, and served as the Managing General Partner of Kodiak Venture Partners, a venture capital firm that is a major shareholder of GreenLight, from February 2006 to February 2014. Dr. Zarur previously served as a senior executive in various companies in the healthcare and clean energy sectors, including as Chief Executive Officer of BioProcessors Corporation, a microscale bioreactor platform developer, from January 2002 to December 2006 and Chief Operating Officer of Starlab NV/SA, a research incubator, from January 1999 to January 2002. Dr. Zarur earned his Doctor of Philosophy degree from the Chemical Engineering Department at the Massachusetts of Technology and a post-graduate certificate in Immunology from Harvard Medical School. Dr. Zarur also holds a Master’s of Science in Engineering Practice from the Chemical Engineering Department at the Massachusetts Institute of Technology and an undergraduate degree from the Universidad Nacional Autónoma de México. We believe Dr. Zarur’s background and track record of leading biopharmaceutical businesses across the discovery, preclinical, and clinical development, commercialization and product life-cycle management states makes him well qualified to serve on the New GreenLight Board.Carole Cobb, M.B.A.
115
GreenLight’s science and innovation. Ms. Cobb’s career has ranged from development and research engineering to managing global manufacturing operations and she was the first female manufacturing Plant Manager at Genencor International, Inc., a research and development company. She was promoted to Vice President of Worldwide Manufacturing in 1997 and from 1999 to 2008, Ms. Cobb was a Senior Vice President of Global Supply at Genencor and continued in that position following its acquisition by Danisco A/S in 2005. In this role, she was responsible for driving strategy, tactics and implementation for Genencor’s global supply chain. Ms. Cobb earned her Master’s in Business Administration from the Finance Department at the University of Rochester and earned two undergraduate degrees, one from the Department of Chemical Engineering and the other from the Department of Biochemistry, and Cell and Molecular Biology both at the State University of New York at Buffalo.
Charu Manocha, M.B.A.
Marta Ortega-Valle, M.B.A
co-founder
of GreenLight, has served as GreenLight’s Chief Business Officer, Human Health since March 2021 and in multiple leading roles at GreenLight since 2009. Ms. Ortega-Valle has led GreenLight’s Human Health business since 2019. Under her leadership, GreenLight is developing multiple mRNA-based drugs, including efforts around rapid pandemic response for vaccine, antibody therapies, and affordable gene therapies. Ms. Ortega-Valle joined Kodiak Venture Partners, a venture capital firm, in 2008 where sheco-founded
GreenLight with Andrey Zarur and James Swartz. Ms. Ortega-Valle started her career in management, strategy, and technology consulting at Accenture plc (NYSE: ACN). During her tenure at Accenture, she led consulting engagements for life sciences and consumer goods multinationals among others industries. In her last role at Accenture Ms. Ortega-Valle was Senior Manager and Director of the Business Intelligence Group in Paris; Since 2019 Ms. Ortega-Valle serves as a member of the board of directors of The Ganeshalab, a Chile-based global biotechscale-up
investment fund for science and technology-based startups. Ms. Ortega-Valle earned her M.B.A. from the Sloan School of Management at MIT, where she was part of the Sloan Fellows Program, and her Master’s degree in Electrical Engineering from the ETSEIB (UPC) Engineering School of Barcelona.Susan Keefe, M.B.A
116
Analytics (2007 to 2013), The Procter & Gamble Company, most recently as Finance Manager (2003 to 2007), as Finance Manager at Lante Corporation (2000 to 2001) and PricewaterhouseCoopers LLP, most recently as Manager, Transaction Services (1996 to 2000). Ms. Keefe earned her B.A. in Business Administration from the University of Iowa and an M.B.A. from the University of Chicago, Booth Graduate School of Business.
David Kennedy.
Amin Khan, Ph.D.
end-to-end
Mark Singleton, Ph.D.
Non-Employee Directors
Charles L. Cooney
117
Robert T. Haslam Professor of Chemical and Biochemical Engineering in the Department of Chemical Engineering at MIT since 2007. In 2015 he became Professor, Emeritus. Dr. Cooney was the founding Faculty Director of the Deshpande Center for Technological Innovation at MIT, from 2002 to 2014. Dr. Cooney also serves as a member of the board of directors of various public and private biotechnology companies, including Elektrofi, a biotechnology company focused on the delivery of biologics to treat diseases, since March 2018; Codiak Biosciences, Inc. (NASDAQ: CDAK), a biotechnology company focused on the development of exosome-based therapeutics, since July 2017, where he also serves as a member of the audit committee and nominating and corporate governance committee; LayerBio, Inc., a biotechnology company focused on sustained-release technology for use with intraocular lenses, since November 2016; Levitronix LLC, a developer of magnetically levitated bearingless motor technology, since January 2016; Innovent Biologics, Inc. (OTCMKTS: IVBXF), a developer of monoclonal antibody drug candidates, since December 2015; and Boyd Technologies, Inc., a developer of membrane products since September 2013. Previously, Dr. Cooney served as a board member at Axcella Health, Inc. (formerly Pronutria Biosciences, Inc.) (NASDAQ: AXLA), a biopharmaceutical company focused on treating diseases and supporting health using endogenous metabolic modulators, from February 2011 to June 2018. Dr. Cooney earned a B.S. in Chemical Engineering from the University of Pennsylvania and an M.S. and Ph.D. in Biochemical Engineering from MIT. We believe that Dr. Cooney’s extensive experience as a researcher and an educator in the biotechnology field and his experience as a director of both public and private biotechnology companies qualify him to serve as a member of the New GreenLight Board.
Ganesh Kishore, Ph.D.
Co-Manager
of MLSCF II (GP) (Labuan), LLP, the General Partner of MLS Capital Fund II. Previously he served as the Chief Executive Officer of Malaysian Life Sciences Capital Fund Ltd., a life sciences venture fund, between April 2007 and June 2015. Additionally, from April 2007 to December 2008, Dr. Kishore served as the Managing Director of Burrill & Company, a life sciences private equity and venture capital firm, in its Venture Group and from 1980 to 2000 Dr. Kishore served as President, Nutrition & Consumer Division, Distinguished Science Fellow and Chief Biotechnologist of Monsanto Company. Dr. Kishore also served as the Chief Technology Officer for Agriculture & Nutrition Platform and Chief Biotechnology Officer of DuPont between 2002 and 2007. Dr. Kishore earned his Ph.D. in Biochemistry from the Indian Institute of Science and received postdoctoral training and was a Robert A. Welch Fellow in Microbiology and Chemistry at the University of Texas at Austin. We believe that Dr. Kishore’s knowledge of the biotechnology sector and his finance experience make him well suited to serve on the New GreenLight Board.Eric O’Brien, M.B.A.
co-founder
and has served as Managing Director of Fall Line Capital, a private equity firm focused on investments in farmland and agricultural technologies, since June 2011. Mr. O’Brien has served on the boards of directors of many public and private companies, including PCH International Company, a custom design manufacturing company, from September 2008 to December 2016; Aquantia Corporation, a manufacturer of high-speed transceivers, from December 2005 to April 2015; Evolv, Inc. (now Cornestone OnDemand), a workforce performance solution analytics company, from January 2008 to November 2014; Partners in School Innovation, anon-profit
service organization, from June 2002 to June 2014; and Lemon, Inc., a mobile wallet developer, from September 2008 to December 2013. Prior to his role at Fall Line Capital, Mr. O’Brien was the Managing Director of Lightspeed Venture Partners, a venture capital firm, from February 2000 to December 2011. Mr. O’Brien earned his M.B.A. from the Stanford University Graduate School of Business and an A.B. in Economics from Harvard College. We believe that Mr. O’Brien is qualified to serve on the New GreenLight118
Board due to his experience and knowledge of GreenLight’s business and his experience in venture capital and finance.
Jennifer E. Pardi
Martha Schlicher, Ph.D
start-up
plant biotechnology company. From February 2016 to December 2019, Dr. Schlicher served as the Vice President of Research and Development of Mallinckrodt Pharmaceutical Company (OTCMKTS: MNKKQ), a manufacturer of specialty pharmaceuticals. Prior to that Dr. Schlicher served in many technical, regulatory, strategy and commercial executive leadership roles at Monsanto, last being as the Vice-President of Sustainability. We believe that Dr. Schlicher’s experience on the GreenLight Board, as well as her experiences running biotechnology companies, make her well qualified to serve on the New GreenLight Board.Matthew Walker, Esq., M.B.A.
non-GMO
commodities market place, since May 2017; and a board member of Farmer Focus-Shenandoah Valley Organic, an organic meat brand, since December 2016. Previously, from August 2011 to February 2014. Mr. Walker was an investment banking associate at Perella Weinberg Partners, a financial services firm, where he focused on merger and acquisitions and restructuring transactions across a range of industries. Prior to that role, from July 2007 to July 2009, Mr. Walker was a securities attorney in the Funds, Regulation, and Equity Derivatives practice at Cadwalader, Wickersham & Taft, LLP, a law firm. Mr. Walker earned his M.B.A. from The University of Chicago Booth School of Business, a J.D. from New York University School of Law, and a B.S. in Mechanical Engineering from the University of Michigan. Mr. Walker is also a member of the Illinois Board of Trustees of The Nature Conservancy. We believe that Mr. Walker’s experience in finance and biotechnology companies, as well as his experience as a member of the GreenLight Board, makes him qualified to serve on the New GreenLight Board.Family Relationships
There are no family relationships among any of the directors and executive officers of New GreenLight.
119
Composition of the Board of Directors Following the Business Combination
In accordance with the terms of the Charter and Bylaws, the New GreenLight Board is divided into three staggered classes of directors, and each director has been assigned to one of the three classes. At each annual meeting of the stockholders, a class of directors will be elected for
a 3-year term
to succeed the directors of the same class whose terms are then expiring. The terms of the directors will expire upon the election and qualification of successor directors at the annual meeting of stockholders to be held during the year 2022 for Class I directors, 2023 for Class II directors and 2024 for Class III directors. Eric O’Brien and Jennifer E. Pardi are Class I directors, Matthew Walker and Martha Schlicher are Class II directors, and Andrey J. Zarur, Charles L. Cooney and Ganesh Kishore are Class III directors.Director Independence
Under the Nasdaq listing standards, a majority of the members of the New GreenLight Board must qualify as “independent,” as affirmatively determined by the New GreenLight Board. Under the rules of Nasdaq, a director will only qualify as an “independent director” if, in the opinion of that company’s board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director. Each individual serving on the New GreenLight Board other than Andrey Zarur qualifies as an independent director under Nasdaq listing standards.
Role of the New GreenLight Board of Directors in Risk Oversight
One of the key functions of the New GreenLight Board is informed oversight of the risk management process. The New GreenLight Board does not have a standing risk management committee, but rather administers this oversight function directly through the New GreenLight Board as a whole, as well as through various standing committees of the New GreenLight Board that address risks inherent in their respective areas of oversight. In particular, the New GreenLight Board is responsible for monitoring and assessing strategic risk exposure and New GreenLight’s audit committee has the responsibility to consider and discuss major financial risk exposures and the steps its management will take to monitor and control such exposures, including guidelines and policies to govern the process by which risk assessment and management is undertaken. The audit committee also monitors compliance with legal and regulatory requirements. New GreenLight’s compensation committee is responsible for overseeing the management of risks relating to executive compensation plans and arrangements. The compensation committee also assesses and monitors whether compensation plans, policies and programs comply with applicable legal and regulatory requirements.
The New GreenLight Board has also adopted a Code of Business Conduct and Ethics for our directors, officers and employees intended to satisfy Nasdaq listing standards and the definition of a “code of ethics” set forth in applicable SEC rules. Our Code of Business Conduct and Ethics is available on our website at www.investors.greenlightbiosciences.com under “Corporate Governance”. The information on our website is not part of this annual report. We intend to make all required disclosures concerning any amendments to, or waivers from, our Code of Business Conduct and Ethics on our website. Any person may request a copy of the Code of Business Conduct and Ethics, at no cost, by writing to us at the following address: GreenLight Biosciences Holdings, PBC, 200 Boston Avenue, Medford, MA 02155, Attention: Corporate Secretary.
Committees of the Board of Directors
The New GreenLight Board has three standing committees: an audit committee, a talent and compensation committee, and a nominating and corporate governance committee.
Audit Committee
The members of New GreenLight’s audit committee are Charles Cooney, Jennifer Pardi and Martha Schlicher. Martha Schlicher serves as the chairperson of the audit committee. Under the Nasdaq listing rules and
120
applicable SEC rules, we are required to have at least three members of the audit committee. The rules of the Nasdaq and
Rule 10A-3 of
the Exchange Act require that the audit committee of a listed company be composed solely of independent directors for audit committee purposes. Each member of the New GreenLight audit committee qualifies as an independent director for audit committee purposes under applicable rules. Each of Charles Cooney, Jennifer Pardi and Martha Schlicher is financially literate, and Martha Schlicher qualifies as an “audit committee financial expert” as defined in applicable SEC rules.The audit committee has the responsibility to, among other things:
| • | select, retain, compensate, evaluate, oversee and, where appropriate, terminate New GreenLight’s independent registered public accounting firm; |
| • | review and approve the scope and plans for the audits and the audit fees and approve all non-audit and tax services to be performed by the independent registered public accounting firm; |
| • | evaluate the independence and qualifications of New GreenLight’s independent registered public accounting firm; |
| • | review New GreenLight’s financial statements, and discuss with management and New GreenLight’s independent registered public accounting firm the results of the annual audit and the quarterly reviews; |
| • | review and discuss with management and New GreenLight’s independent registered public accounting firm the quality and adequacy of New GreenLight’s internal controls and New GreenLight’s disclosure controls and procedures; |
| • | discuss with management New GreenLight’s procedures regarding the presentation of New GreenLight’s financial information, and review earnings press releases and guidance; |
| • | oversee the design, implementation and performance of New GreenLight’s internal audit function, if any; |
| • | set hiring policies with regard to the hiring of employees and former employees of New GreenLight’s independent registered public accounting firm and oversee compliance with such policies; |
| • | review, approve and monitor related party transactions; |
| • | review and monitor compliance with New GreenLight’s Code of Business Conduct and Ethics and consider questions of actual or possible conflicts of interest of New GreenLight’s directors and officers; |
| • | adopt and oversee procedures to address complaints regarding accounting, internal accounting controls and auditing matters, including confidential, anonymous submissions by New GreenLight’s employees of concerns regarding questionable accounting or auditing matters; |
| • | review and discuss with management and New GreenLight’s independent registered public accounting firm the adequacy and effectiveness of New GreenLight’s legal, regulatory and ethical compliance programs; and |
| • | review and discuss with management and New GreenLight’s independent registered public accounting firm New GreenLight’s guidelines and policies to identify, monitor and address enterprise risks. |
New GreenLight’s audit committee operates under a written charter that satisfies the applicable rules and regulations of the SEC and the listing standards of Nasdaq.
Compensation Committee
New GreenLight’s compensation committee consists of at least three members of the New GreenLight Board, all of whom are independent directors. The members of the compensation committee are Ganesh Kishore, Eric O’Brien, Martha Schlicher and Matthew Walker. Ganesh Kishore serves as the chairperson of the compensation committee.
121
The New GreenLight compensation committee has the responsibility to, among other things:
| • | review and approve or recommend to the New GreenLight Board for approval the compensation for New GreenLight’s executive officers, including New GreenLight’s chief executive officer; |
| • | review, approve and administer New GreenLight’s employee benefit and equity incentive plans; |
| • | advise the New GreenLight Board on stockholder proposals related to executive compensation matters; |
| • | establish and review the compensation plans and programs of New GreenLight’s employees, and ensure that they are consistent with New GreenLight’s general compensation strategy; |
| • | oversee the management of risks relating to executive compensation plans and arrangements; |
| • | monitor compliance with any stock ownership guidelines; |
| • | approve the creation or revision of any clawback policy; |
| • | review and approve or recommend to the New GreenLight Board for approval non-employee director compensation; and |
| • | review executive compensation disclosure in New GreenLight’s SEC filings and prepare the compensation committee report required to be included in New GreenLight’s annual proxy statement. |
New GreenLight’s compensation committee operates under a written charter that satisfies the applicable rules and regulations of the SEC and the listing standards of Nasdaq.
Compensation Committee Interlocks and Insider Participation
No member of the compensation committee was at any time during 2021, or at any other time, one of our officers or employees. None of our executive officers has served as a director or member of a compensation committee (or other committee serving an equivalent function) of any entity, one of whose executive officers served as a member of our board of directors or member of our compensation committee.
Nominating and Corporate Governance Committee
New GreenLight’s nominating and corporate governance committee consists of at least two members of the New GreenLight Board, all of whom are independent directors. The members of the nominating and corporate governance committee are Ganesh Kishore, Jennifer Pardi and Matthew Walker. Matthew Walker serves as the chairperson of the nominating and corporate governance committee.
New GreenLight’s nominating and corporate governance committee has the responsibility to, among other things:
| • | review, assess and make recommendations to the New GreenLight Board regarding desired qualifications, expertise and characteristics sought of board members; |
| • | identify, evaluate, select or make recommendations to the New GreenLight Board regarding nominees for election to the New GreenLight Board; |
| • | develop policies and procedures for considering stockholder nominees for election to the New GreenLight Board; |
| • | review New GreenLight’s succession planning process for New GreenLight’s chief executive officer and any other members of New GreenLight’s executive management team; |
| • | review and make recommendations to the New GreenLight Board regarding the composition, organization and governance the New GreenLight Board and its committees; |
122
| • | review and make recommendations to the New GreenLight Board regarding New GreenLight’s corporate governance guidelines and corporate governance framework; |
| • | oversee director orientation for new directors and continuing education for New GreenLight’s directors; |
| • | oversee New GreenLight’s ESG programs and related disclosures and communications; |
| • | oversee the evaluation of the performance of the New GreenLight Board and its committees; and |
| • | administer policies and procedures for communications with the non-management members of the New GreenLight Board. |
New GreenLight’s nominating and corporate governance committee operates under a written charter that satisfies the applicable rules and regulations of the SEC and the listing standards of Nasdaq.
Director Nominations
New GreenLight’s nominating and corporate governance committee will consider director candidates recommended for nomination by New GreenLight’s stockholders during such times as they are seeking proposed nominees to stand for election at the next annual meeting of stockholders (or, if applicable, a special meeting of stockholders). New GreenLight’s stockholders that wish to nominate a director for election to New GreenLi board of directors should follow the procedures set forth in New GreenLight’s bylaws.
New GreenLight’s nominating and corporate governance committee has not formally established any specific, minimum qualifications that must be met or skills that are necessary for directors to possess. In general, in identifying and evaluating nominees for director, the nominating and corporate governance committee will consider business experience, expertise and acumen, knowledge of New GreenLight’s business and industry, diversity of professional experience, educational background, integrity, professional reputation, independence, wisdom, and the ability to represent the best interests of New GreenLight’s stockholders.
Director Compensation
The New GreenLight Board with the recommendation of New GreenLight’s compensation committee will determine whether and how to change the annual compensation to be paid to the members of the New GreenLight Board in order to include an equity component in board compensation.
Executive Compensation
New GreenLight intends to develop an executive compensation program that is designed to align compensation with New GreenLight’s business objectives and the creation of stockholder value, while enabling New GreenLight to attract, motivate and retain individuals who contribute to the long-term success of New GreenLight.
Decisions on the executive compensation program will be made by New GreenLight’s compensation committee.
Delinquent Section 16(a) Reports
Section 16(a) of the Exchange Act requires executive officers and directors of New GreenLight, and persons who own more than ten percent of any publicly traded class of equity securities of New GreenLight, to file reports of ownership and changes in ownership of equity securities of New GreenLight with the SEC. Officers, directors, andstockholders are required by the SEC’s regulations to furnish New GreenLight with copies of all Section 16(a) forms that they file.
greater-than-ten-percent
123
Based solely upon a review of Forms 3 and Forms 4 furnished to New GreenLight during the most recent fiscal year, and Forms 5 with respect to its most recent fiscal year, we believe that all such forms required to be filed pursuant to Section 16(a) of the Exchange Act were timely filed by the officers, directors, and security holders required to file the same with respect to the fiscal year ended December 31, 2021.
ITEM 11. Executive Compensation
Executive Compensation
Unless the context otherwise requires, any reference in this section of this annual report to “GreenLight,” “we,” “us” or “our” refers to GreenLight and its consolidated subsidiaries prior to the consummation of the Business Combination and to New GreenLight and its consolidated subsidiaries following the Business Combination.
GreenLight aims to pay competitively to attract, develop and retain highly talented employees. We provide rewards for high performance and critical skills and design compensation programs and structures to provide transparency around what is expected, encourage and reward delivery of annual objectives that are aligned with our stockholders’ long-term interests, and ultimately, support the achievement of GreenLight’s business strategy. GreenLight’s executive compensation program consists primarily of base salaries, an annual performance-based bonus program, and our equity-based incentive compensation program under the GreenLight 2012 Stock Incentive Plan (the “
GreenLight 2012 equity plan
”). Upon the consummation of the Business Combination, New GreenLight adopted the New GreenLight Equity Plan and the New GreenLight ESPP, which are described in more detail below, and terminated the GreenLight 2012 equity plan.This section provides an overview of GreenLight’s executive compensation programs, including a narrative description of the material factors necessary to understand the information disclosed in the summary compensation table below.
At December 31, 2021, GreenLight’s named executive officers were:
| • | Dr. Andrey Zarur, President and Chief Executive Officer |
| • | Carole B. Cobb, Chief Operating Officer |
| • | Susan E. Keefe, Chief Financial Officer |
124
Summary Compensation Table for the year ended December 31, 2021
The following table provides information regarding the compensation earned by GreenLight’s named executive officers for the years ended December 31, 2021. During the period prior to the consummation of the Business Combination on February 2, 2022, ENVI did not pay any compensation to its executive officers.
Name and principal position | Year | Salary ($) | Option awards ($) (1) | Non-equity incentive plan compensation ($) (2) | All other compensation ($) (3) | Total ($) | ||||||||||||||||||
Dr. Andrey Zarur President and Chief Executive Officer | 2021 | 500,000 | — | 227,500 | 279 | 727,779 | ||||||||||||||||||
| 2020 | 450,000 | 1,146,000 | (4) | 180,000 | 285 | 1,776,285 | ||||||||||||||||||
Carole B. Cobb Chief Operating Officer | 2021 | 425,000 | — | 154,700 | 279 | 579,979 | ||||||||||||||||||
| 2020 | 350,000 | 196,000 | 105,000 | 285 | 651,285 | |||||||||||||||||||
Susan E. Keefe Chief Financial Officer | 2021 | 380,000 | — | 172,900 | 279 | 553,179 | ||||||||||||||||||
| 2020 | 325,000 | 340,000 | 97,500 | 285 | 762,785 | |||||||||||||||||||
| (1) | In accordance with SEC rules, this column reflects the aggregate grant date fair value of the stock option awards granted during the year ended December 31, 2021, computed in accordance with FASB ASC 718. These amounts do not reflect the actual economic value that will be realized by the named executive officer upon the vesting of the stock options, the exercise of the stock options, or the sale of the shares underlying such stock options. There were no stock options granted in the year ended December 31, 2021. For a description of the determination of the fair value of the stock option awards, see the GreenLight MD&A under the subheading “ — Critical Accounting Policies and Significant Judgments and Estimates — Determination of the Fair Value of Common Stock” Outstanding Equity Awards at December 31, 2021, —Employment Arrangements with Officers —GreenLight 2012 Stock Incentive Plan |
| (2) | These amounts represent performance-based cash bonuses based upon the achievement of goals for 2020 and 2021. Performance-based goals for 2020 were earned in 2020 and paid in 2021. The majority of the performance-based goals for 2021 were earned in 2021 and paid in 2022. GreenLight’s bonus program is more fully described below under the section titled “ GreenLight Executive Compensation—Non-Equity Incentive Plan Compensation |
| (3) | These amounts represent life insurance premiums paid by GreenLight for the benefit of the named executive officers. |
| (4) | In the year ended December 31, 2020, Dr. Zarur received two stock option awards, one of which is a performance-based award and one of which is a service-based award. At the date of grant, achievement of the conditions in the performance-based award was deemed not probable and, accordingly, the grant date fair value of the award was zero based upon the probable outcome of such conditions. Assuming achievement of the highest level of performance, the performance-based award would have had a grant date fair value of $162,681. In December 2021, the GreenLight Board voted to extend the length of time to allow for the performance vesting to occur by March 31, 2022. The fair value of the award, as modified, was $2,092,472 as of the modification date. Accordingly, the value reflected in the table represents only the grant date fair value of the service-based award. |
Base Salaries
Base salary is a fixed element within a total compensation package intended to attract and retain the talent necessary to successfully manage GreenLight’s business and execute its business strategies. Base salaries for
125
GreenLight’s named executive officers are established based on the scope of their responsibilities, taking into account relevant experience, internal pay equity, tenure, GreenLight’s ability to replace the individual, and other factors deemed relevant.
Base salaries for the named executive officers as of January 1, 2022 and 2021 were increased as follows:
Name | 2022 Base Salary | 2021 Base Salary | ||||||
Dr. Andrey Zarur | $ | 575,000 | $ | 500,000 | ||||
Carole B. Cobb | $ | 440,000 | $ | 425,000 | ||||
Susan E. Keefe | $ | 415,000 | $ | 380,000 | ||||
Non-Equity Incentive
Plan CompensationAt the beginning of 2021 and 2020, the GreenLight Board set company goals for 2021 and 2020 with the objective of paying cash bonuses in 2022 and 2021 based on achievement of the 2021 and 2020 goals, respectively. Company goals consisted of both corporate development and product development goals to be measured and granted at the sole discretion of the GreenLight Board in 2022 and 2021.
Based on GreenLight’s performance against the company goals for 2021, the GreenLight Board determined to award each of Dr. Zarur and Ms. Cobb 91% of his or her target bonus amount for that year and to award Ms. Keefe approximately 114% of her target bonus amount for that year. Based on GreenLight’s performance against the company goals for 2020, the GreenLight Board determined to award each of the named executive officers 100% of their target bonus amounts for that year.
The amounts in the Summary Compensation Table under the
column “Non-equity incentive
plan compensation” are based on each named executive officer’s individual target bonus amount multiplied by the achievement percentage set by the GreenLight Board. For 2020, the target bonus amounts for Dr. Zarur, Ms. Cobb and Ms. Keefe, as a percentage of base salary, were 40%, 30% and 30%, respectively, and were 100% based on achievement of company goals. For 2021, the target bonus amounts for Dr. Zarur, Ms. Cobb and Ms. Keefe, as a percentage of base salary, were 50%, 40% and 40%, respectively.The target bonus amounts for the named executive officers under the employee bonus program, as a percentage of base salary, were increased as follows as of January 1, 2022 and 2021:
Name | 2022 Target Bonus Amount | 2021 Target Bonus Amount | ||||||
Dr. Andrey Zarur | 55 | % | 50 | % | ||||
Carole B. Cobb | 45 | % | 40 | % | ||||
Susan E. Keefe | 45 | % | 40 | % | ||||
Equity Based Incentive Awards
GreenLight’s equity-based incentive awards are designed to more closely align GreenLight’s interests and those of GreenLight’s stockholders with those of GreenLight’s employees and consultants, including GreenLight’s named executive officers. The GreenLight Board is responsible for approving equity grants to GreenLight’s employees and consultants, including GreenLight’s named executive officers.
All options are granted with an exercise price per share that is no less than the fair market value of a share of GreenLight Common Stock on the date of grant of such award. GreenLight’s stock option awards generally vest over a four-year period and may be subject to acceleration of vesting and exercisability under certain termination and change in control events. See “—.”
Outstanding Equity Awards at December
31, 2021
126
Outstanding Equity Awards at December 31, 2021
The following table provides information regarding equity awards granted to GreenLight’s named executive officers that remain outstanding as of December 31, 2021. The number of securities underlying unexercised options as of December 31, 2021 represent shares of GreenLight Common Stock, and neither such numbers nor the associated exercise prices give effect to the conversion of such options upon the consummation of the Business Combination on February 2, 2022 into options to acquire shares of New GreenLight Common Stock at the Exchange Ratio of 0.6656 shares of New GreenLight Common Stock for each share of GreenLight Common Stock.
Option awards (1) | ||||||||||||||||||||||||
Name | Grant date | Number of securities underlying unexercised options (#) exercisable | Number of securities underlying unexercised options (#) unexercisable | Equity incentive plan awards: Number of securities underlying unexercised unearned options (#) | Option exercise price ($) (2) | Option expiration date | ||||||||||||||||||
Dr. Andrey Zarur | 10/16/2015 | 105,648 | — | — | $ | 0.23 | 07/22/2025 | |||||||||||||||||
| 09/13/2018 | (3) | 1,182,963 | 208,759 | — | $ | 0.22 | 09/13/2028 | |||||||||||||||||
| 12/29/2019 | (3) | 1,593,412 | 2,390,121 | — | $ | 0.33 | 12/29/2029 | |||||||||||||||||
| 12/01/2020 | (4) | — | — | 439,678 | $ | 0.65 | 12/01/2030 | |||||||||||||||||
| 12/01/2020 | (5) | 716,250 | 2,148,750 | — | $ | 0.65 | 12/01/2030 | |||||||||||||||||
Carole B. Cobb | 07/22/2015 | (3) | 642,657 | — | — | $ | 0.23 | 07/22/2025 | ||||||||||||||||
| 10/21/2016 | (3) | 65,900 | — | — | $ | 0.23 | 10/21/2026 | |||||||||||||||||
| 02/14/2018 | (3) | 586,500 | 103,500 | — | $ | 0.22 | 02/14/2028 | |||||||||||||||||
| 09/13/2018 | (3) | 202,242 | 79,957 | — | $ | 0.22 | 09/13/2028 | |||||||||||||||||
| 12/29/2019 | (3) | 305,015 | 457,527 | — | $ | 0.33 | 12/29/2029 | |||||||||||||||||
| 12/01/2020 | (5) | 122,500 | 367,500 | — | $ | 0.65 | 12/01/2030 | |||||||||||||||||
Susan E. Keefe | 05/10/2019 | (3) | 366,937 | 366,937 | — | $ | 0.33 | 05/10/2029 | ||||||||||||||||
| 12/01/2020 | (5) | 212,500 | 637,500 | — | $ | 0.65 | 12/01/2030 | |||||||||||||||||
| (1) | All of the outstanding stock option awards were granted under and subject to the terms of the GreenLight 2012 equity plan, described below under “ — GreenLight 2012 Stock Incentive Plan |
| (2) | The stock option awards were granted with a per share exercise price equal to the fair market value of one share of GreenLight Common Stock on the date of grant, as determined in good faith by the GreenLight Board. |
| (3) | The stock option award vests as to 20% of the total number of shares subject to the award on the first anniversary of the vesting start date (which in some cases precedes the date of grant), and the remainder vests in 48 equal monthly installments thereafter. |
| (4) | The stock option award is subject to performance-based vesting conditions. The award will vest as described in footnote (5) below, provided that GreenLight consummates a specified new investment (which for this purpose includes the Business Combination) by March 31, 2022. |
| (5) | The stock option award vests as to 25% of the total number of shares subject to the award on the first anniversary of the vesting start date (which in some cases precedes the date of grant), and the remainder vests in 36 equal monthly installments thereafter. |
Employment Arrangements with Officers
Dr. Andrey Zarur
GreenLight entered into an Amended and Restated Employment Agreement with Dr. Andrey Zarur, dated May 25, 2015 (the “
Zarur Agreement
”) which governs Dr. Zarur’s role as President and Chief Executive Officer of GreenLight. Dr. Zarur’s employment under the Zarur Agreementis at-will and
will continue until terminated at any time by either party. Pursuant to the Zarur Agreement, Dr. Zarur was initially entitled to receive a127
specified annual base salary for 2015. Dr. Zarur’s salary was most recently increased to $575,000 effective as of January 1, 2022. Dr. Zarur was also initially eligible to receive a discretionary annual bonus equal to up to 40% of his base salary (which was increased to 55% effective as of January 1, 2022) and options to purchase shares of GreenLight Common Stock (as detailed in the Zarur Agreement), subject to vesting over five years. The Zarur Agreement provides for acceleration of vesting of any unvested options upon termination without Cause or for Good Reason (each, as defined in the Zarur Agreement) in connection with a Change of Control Event (as defined in the Zarur Agreement). Additional information regarding Dr. Zarur’s outstanding option awards can be found under the section titled “.” Dr. Zarur is also entitled to participate in GreenLight’s employee benefit plans and
GreenLight Executive Compensation —
Outstanding Equity Awards at December
31, 2021
paid time-off policies.
If Dr. Zarur’s employment is terminated by GreenLight without Cause (as defined in the Zarur Agreement) or due to illness, accident or disability prohibiting him from performing his duties for three months in
a 12-month period,
or by Dr. Zarur for Good Reason (as defined in the Zarur Agreement), then Dr. Zarur will be entitled to severance equal to one year’s salary payable over thesucceeding 12-month period.
If payments owed to Dr. Zarur at the time of termination would trigger the acceleration or increase of tax payable under Section 409A of the Code, GreenLight agreed to defer the commencement of such payments until the date that is at least six months following such termination, at which time GreenLight will pay Dr. Zarur alump-sum
payment equal to the cumulative amounts that would have otherwise been previously paid to Dr. Zarur during such period of deferral. Dr. Zarur’s right to severance is contingent upon his execution of a general release of claims in favor of GreenLight and his continued compliance withhis non-competition and
confidentiality agreement with GreenLight.Carole B. Cobb
GreenLight entered into an offer letter with Carole B. Cobb, dated July 21, 2015, that provides for
her at-will employment
as GreenLight’s Senior Vice President of Operations. Ms. Cobb was subsequently named our Chief Operations Officer. The offer letter initially provided for Ms. Cobb to receive a specified annual base salary for 2015. Ms. Cobb’s salary was most recently increased to $440,000 effective as of January 1, 2022. Ms. Cobb was also initially eligible to receive a discretionary annual bonus equal to up to 30% of her annual base salary (which was increased to 45% effective as of January 1, 2022) and an option to purchase shares of GreenLight Common Stock, subject to vesting over five years. The offer letter provides for acceleration of vesting of any unvested options in the event of termination without Cause or by Ms. Cobb for Good Reason (each, as defined in the offer letter) in connection with a Change of Control Event (as defined in the offer letter).If Ms. Cobb’s employment is terminated by GreenLight without Cause (as defined in the offer letter) or by Ms. Cobb for Good Reason (as defined in the offer letter), she will be entitled to severance equal to three months’ salary, payable in a lump sum within seven days of her termination of employment. Ms. Cobb’s right to severance is contingent upon her execution of a general release of claims in favor of GreenLight. Ms. Cobb is also entitled to participate in GreenLight’s employee benefit plans and paid-time off policies.
Susan E. Keefe
GreenLight entered into an offer letter with Susan E. Keefe, dated May 1, 2019, that provides for
her at-will employment
as GreenLight’s Chief Financial Officer. The offer letter provided for Ms. Keefe to receive a specified annual base salary for 2019. Ms. Keefe’s salary was most recently increased to $415,000 effective as of January 1, 2022. Ms. Keefe was also initially eligible to receive a discretionary annual bonus equal to up to 30% of her annual base salary (which was increased to 45% effective as of January 1, 2022), and an option to purchase shares of GreenLight Common Stock subject to vesting over five years. The offer letter provides for acceleration of vesting of any unvested options upon termination without Cause (as defined in the offer letter) in connection with a Change in Control Transaction (as defined in the offer letter). Pursuant to the offer letter, Ms. Keefe was also eligible to receive (and did receive) an additional option grant subject to128
performance milestones. If Ms. Keefe’s employment is terminated by GreenLight without Cause (as defined in the offer letter) or by Ms. Keefe for good reason (as defined in the offer letter), she will be entitled to severance equal to six months’ salary payable over the six months following such termination. Ms. Keefe is also entitled to participate in GreenLight’s employee benefit plans and
paid time-off policies.
Potential Payments Upon Termination or Change of Control
Regardless of the manner in which a named executive officer’s service terminates, that named executive officer is entitled to receive amounts earned during his or her term of service, including unpaid salary and unused vacation, as applicable.
Each named executive officer holds stock options granted subject to the general terms of the GreenLight 2012 equity plan. A description of the terminationin the GreenLight 2012 equity plan and applicable to the stock options granted to GreenLight’s named executive officers is provided below under.”
and change-in-control provisions
“—
GreenLight 2012 Stock Incentive Plan
For additional information regarding potential payments and acceleration of stock options upon termination or change of control, see” above.
“—
Employment Arrangements with Officers
Benefits and Perquisites
GreenLight provides benefits to its named executive officers on the same basis as provided to all of its employees, including health, dental and vision insurance; life insurance; accidental death and dismemberment
insurance; short-and long-term
disability insurance; anda tax-qualified Section 401(k)
plan for which no match is provided by GreenLight. GreenLight provides an enhanced life insurance benefit to its executive officers but does not otherwise maintain any executive-specific benefit or perquisite programs.Retirement Benefits
GreenLight maintains a 401(k) retirement savings plan, for the benefit of employees, including its named executive officers, who satisfy certain eligibility requirements. The 401(k) plan provides eligible employees with an opportunity to save for retirement onbasis, through contributions to the 401(k) plan. All of a participant’s contributions into the 401(k) plan are 100% vested when contributed. The 401(k) plan is intended to qualify under Sections 401(a) and 501(a) of the Code. As
a tax-advantaged basis.
Under the 401(k) plan, eligible employees may elect to defer a portion of their compensation, within the limits prescribed by the Code and the applicable limits under the 401(k) plan, ona pre-tax or after-tax (Roth)
a tax-qualified retirement
plan, pre-tax contributions
to the 401(k) plan and earnings onthose pre-tax contributions
are not taxable to the employees until distributed from the 401(k) plan, and earnings on Roth contributions are not taxable when distributed from the 401(k) plan. GreenLight does not provide a match for participants’ elective contributions to the 401(k) plan, nor does GreenLight provide to employees, including its named executive officers, any other retirement benefits, including without limitationany tax-qualified defined
benefit plans, supplemental executive retirement plans and nonqualified defined contribution plans.GreenLight 2012 Stock Incentive Plan
The GreenLight 2012 equity plan provided for the grant of equity-based awards, denominated in GreenLight Common Stock, including incentive
stock options, non-statutory stock options
and restricted stock awards. We will not make any new awards under the GreenLight 2012 equity plan. Pursuant to the terms of the Business Combination Agreement, upon consummation of the Business Combination, all outstanding equity awards under the GreenLight 2012 equity plan were converted into comparable equity awards governed by the New GreenLight Equity Plan, which is currently administered by the Compensation Committee of the New GreenLight Board. The material features of the GreenLight 2012 equity plan are summarized below.129
General
Purposes
Administration
Source of shares
Eligibility
Amendment or termination of our stock incentive plan
Options
as non-statutory
stock options. We may grantnon-statutory
stock options to our employees, directors, officers, consultants or advisors in the discretion of the GreenLight Board. Incentive stock options will only be granted to our employees.The exercise price of each incentive stock
option and non-statutory stock option
may not be less than 100% of the fair market value of GreenLight Common Stock on the date of grant. If we grant incentive stock options to any 10% stockholder, the exercise price may not be less than 110% of the fair market value of GreenLight Common Stock on the date of grant. The term of each option may not exceed 10 years from the date of grant, except that the term of any incentive stock option granted to any 10% stockholder may not exceed five years from the date of grant. At the time of grant of the award, the GreenLight Board determines at what time or times each option may be exercised and the period of time, if any, after death, disability or termination of employment during which options may be exercised. Options may be made exercisable in installments. The vesting and exercisability of options may be accelerated by the GreenLight Board.In general, an optionee may pay the exercise price of an option by cash or check or, if so provided in the applicable option agreement and with the written consent of the GreenLight Board, by tendering GreenLight Common Stock having a fair market value equal to the aggregate exercise price of the options being exercised, by a personal recourse note issued by the optionee in a principal amount equal to the aggregate exercise price of the options being exercised, by a “cashless exercise” through a broker supported by irrevocable instructions to the broker to deliver sufficient funds to pay the applicable exercise price, by reducing the number of shares
130
otherwise issuable to the optionee upon exercise of the option by a number of shares having a fair market value equal to the aggregate exercise price of the options being exercised, or by any combination of these methods of payment.
Incentive stock options granted under the GreenLight 2012 equity plan may not be transferred or assigned other than by will or under applicable laws of descent and distribution. The GreenLight Board may determine the extent to
which a non-statutory option shall
be transferable.Restricted stock awards
achievement of pre-established performance goals
and objectives.Adjustments upon changes in capitalization
Effect of a change in control
| • | provide that such stock options will be assumed, or equivalent stock options substituted, by the acquiring or succeeding corporation (or an affiliate thereof); |
| • | upon written notice to the optionees, provide that all unexercised stock options will terminate immediately prior to the consummation of the change in control transaction unless exercised by the optionee to the extent otherwise then exercisable within a specified period following the date of such notice; |
| • | upon written notice to the grantees, provide that all unvested shares of restricted stock will be repurchased at cost; |
| • | make or provide for a cash payment to the optionees equal to the difference between (x) the fair market value of the per share consideration (whether cash, securities or other property or any combination thereof) the holder of a GreenLight Common Share will receive upon consummation of the change in control transaction times the number of GreenLight Common Stock subject to outstanding vested stock options (to the extent then exercisable at prices not equal to or in excess of such per share consideration) and (y) the aggregate exercise price of such outstanding vested stock options, in exchange for the termination of such stock options; or |
| • | provide that all or any outstanding stock options will become exercisable and all or any outstanding restricted stock awards will vest in part or in full immediately prior to the change in control transaction. To the extent that any stock options are exercisable at a price equal to or in excess of the per share consideration in the change in control transaction, the GreenLight Board may provide that such stock options will terminate immediately upon the consummation of the change in control transaction without any payment being made to the holders of such stock options. |
For additional information regarding potential acceleration of vesting of stock options held by our named executive officers upon termination or change of control, see “” above.
—Employment Arrangements with Officers
131
New GreenLight 2022 Equity and Incentive Plan
The following is a summary of the material features of the New GreenLight Equity Plan. This summary is qualified in its entirety by the full text of the New GreenLight Equity Plan, a copy of which is filed as an exhibit to this annual report.
The New GreenLight Equity Plan has been adopted by the Board: and approved by our stockholders. The New GreenLight Equity Plan became effective immediately prior to the consummation of the Business Combination (the “
Equity Plan Effective Date
”). The New GreenLight Equity Plan allows New GreenLight to make equity and equity-based incentive awards, as well as cash awards, to employees, directors and consultants. The Board anticipates that providing such persons with a direct stake in New GreenLight will assure a closer alignment of the interests of such individuals with those of New GreenLight and its stockholders, thereby stimulating their efforts on New GreenLight’s behalf and strengthening their desire to remain with New GreenLight.Purposes of the New GreenLight Equity Plan
The purposes of the New GreenLight Equity Plan are to attract and retain personnel for positions with New GreenLight or any subsidiary of New GreenLight; to provide additional incentive to employees, directors, and consultants; and to promote the success of New GreenLight’s business. These incentives will be provided through the grant of stock options, stock appreciation rights, restricted stock unrestricted stock, restricted stock units, dividend equivalent rights, and cash awards as the administrator of the New GreenLight Equity Plan may determine.
Eligibility
As of December 31, approximately 295 individuals are eligible to participate in the New GreenLight Equity Plan, which includes approximately
6 non-employee
directors, 8 officers and 281 employees who are not officers. In addition, our consultants are also generally eligible to participate in the New GreenLight Equity Plan.The awards that are to be granted to any participant or group of participants are indeterminable at the date of this annual report because participation and the types of awards that may be granted under the New GreenLight Equity Plan are subject to the discretion of the plan administrator. Consequently, no new plan benefits table is included in this annual report.
No awards may be granted under the New GreenLight Equity Plan after the date that is ten years from the New GreenLight Equity Plan Effective Date, and awards of incentive stock options may not be granted after the date that is ten years from the date the New GreenLight Equity Plan was approved by the Board.
Authorized Shares
New GreenLight has initially reserved 31,750,000 shares of New GreenLight Common Stock for issuance under the New GreenLight Equity Plan (the “
Initial Limit
”), and shares subject to the Rollover Options count against this limit. The New GreenLight Equity Plan provides that the number of shares of New GreenLight Common Stock reserved and available for issuance under the New GreenLight Equity Plan will automatically increase each January 1, beginning on January 1, 2023 and on each January 1 thereafter, by 4% of the outstanding number of shares of New GreenLight Common Stock on the immediately preceding December 31, or such lesser amount as determined by the plan administrator (the “Annual Increase
”). This limit is subject to adjustment in the event of a reorganization, recapitalization, reclassification, stock split, stock dividend, reverse stock split or other similar change in New GreenLight’s capitalization. The maximum aggregate number of shares of New GreenLight Common Stock that may be issued upon exercise of incentive stock options under the New GreenLight Equity Plan may not exceed the Initial Limit cumulatively increased on January 1, 2023 and on132
each January 1 thereafter by the lesser of the Annual Increase or a number of shares of New GreenLight Common Stock equal to twice the Initial Limit. Shares underlying any awards under the New GreenLight Equity Plan that are forfeited, cancelled, held back upon exercise of an option or settlement of an award to cover the exercise price or tax withholding, reacquired by New GreenLight prior to vesting, satisfied without the issuance of stock or otherwise terminated (other than by exercise) will be added back to the shares available for issuance under the New GreenLight Equity Plan and, to the extent permitted under Section 422 of the Code and the regulations promulgated thereunder, the shares may be issued as incentive stock options. In addition, to the extent consistent with the requirements of Section 422 of the Code, awards granted or stock issued upon assumption of, or in substitution or exchange for, awards previously granted by an entity that New GreenLight acquires or merges with or into, shall not reduce the shares available for issuance under the New GreenLight Equity Plan, nor will the shares underlying such awards be added back to the shares available for issuance under the New GreenLight Equity Plan in the event of any forfeiture, cancelation, reacquisition, expiration, termination, cash settlement or
non-issuance
of such shares.The New GreenLight Equity Plan contains a limitation whereby the value of all awards under the New GreenLight Equity Plan and all other cash compensation paid by New GreenLight to any
non-employee
director may not exceed $375,000 in any calendar year, except that the limit will be $500,000 for the first calendar year anon-employee
director is initially appointed to the New GreenLight Board. The foregoing limitation will be calculated without regard to amounts paid to anynon-employee
director (including retirement benefits and severance payments) in respect of any services provided in any capacity (including employee or consultant) other than as anon-employee
director. The New GreenLight Board may make exceptions to this limit for anon-executive
chair of the New GreenLight Board with the approval of a majority of the disinterested directors.Plan Administration
The New GreenLight Equity Plan will be administered by the compensation committee of the New GreenLight Board, the New GreenLight Board or another board committee pursuant to the terms of the New GreenLight Equity Plan. The plan administrator, which initially is the compensation committee of the New GreenLight Board, will have full power to select, from among the individuals eligible for awards, the individuals to whom awards will be granted, to make awards to participants, and to determine the specific terms and conditions of each award, subject to the provisions of the New GreenLight Equity Plan. The plan administrator may, without the approval of New GreenLight’s stockholders, reduce the exercise price of any outstanding stock option or stock appreciation right, effect the repricing of such awards through cancellation and
re-grants,
or cancel such awards in exchange for cash or other awards. The plan administrator’s determinations under the New GreenLight Equity Plan need not be uniform. The plan administrator may delegate to one or more officers the authority to grant stock options and other awards to employees who are not subject to the reporting and other provisions of Section 16 of the Exchange Act, subject to certain limitations and guidelines. Persons eligible to participate in the New GreenLight Equity Plan are the directors, officers, employees and consultants of New GreenLight and its affiliates as selected from time to time by the plan administrator in its discretion.The New GreenLight Equity Plan requires the plan administrator to make appropriate adjustments to the number of shares of New GreenLight Common Stock that are subject to the New GreenLight Equity Plan, to certain limits in the New GreenLight Equity Plan, and to any outstanding awards to reflect stock dividends, stock splits, extraordinary cash dividends and similar events.
Stock Options
The New GreenLight Equity Plan permits the granting of both options to purchase shares of New GreenLight Common Stock intended to qualify as incentive stock options under Section 422 of the Code and options that do not so qualify. Options granted under the New GreenLight Equity Plan will be
non-qualified
options if they fail to qualify as incentive stock options or exceed the annual limit on incentive stock options. Incentive stock options may only be granted to employees of New GreenLight and its subsidiaries.Non-qualified
133
options may be granted to any persons eligible to receive awards under the New GreenLight Equity Plan. The option exercise price of each option will be determined by the plan administrator but generally may not be less than 100% of the fair market value of New GreenLight Common Stock on the date of grant or, in the case of an incentive stock option granted to a ten percent stockholder, 110% of such share’s fair market value on the date of grant. The term of each option will be fixed by the plan administrator and may not exceed ten years from the date of grant, subject to limited exceptions as described in the New GreenLight Equity Plan. The plan administrator will determine at what time or times each option may be exercised, including the ability to accelerate the vesting of such options.
Upon exercise of an option, the option exercise price must be paid in full either in cash, by certified or bank check or other instrument acceptable to the plan administrator or by delivery (or attestation to the ownership) of shares of New GreenLight Common Stock that are beneficially owned by the optionee free of restrictions or were purchased in the open market. The exercise price may also be delivered by a broker pursuant to irrevocable instructions to the broker from the optionee. In addition, the plan administrator may permit options to be exercised using a “net exercise” arrangement that reduces the number of shares issued to the optionee by the largest whole number of shares with a fair market value that does not exceed the aggregate exercise price.
Stock Appreciation Rights
The plan administrator may award stock appreciation rights subject to such conditions and restrictions as it may determine. Stock appreciation rights entitle the recipient to receive shares of New GreenLight Common Stock, or cash to the extent provided for in an award agreement, equal to the value of the appreciation in New GreenLight Common Stock price over the exercise price. The exercise price generally may not be less than 100% of the fair market value of New GreenLight Common Stock on the date of grant. The term of each stock appreciation right will be fixed by the plan administrator and may not exceed ten years from the date of grant, subject to limited exceptions as described in the New GreenLight Equity Plan. The plan administrator will determine at what time or times each stock appreciation right may be exercised.
Restricted Stock, Restricted Stock Units, Unrestricted Stock, Dividend Equivalent Rights
The plan administrator may award restricted shares of New GreenLight Common Stock and restricted stock units subject to such conditions and restrictions as it may determine. These conditions and restrictions may include the achievement of certain performance goals and/or continued employment through a specified vesting period. The plan administrator may also grant shares of New GreenLight Common Stock that are free from any restrictions under the New GreenLight Equity Plan. Unrestricted stock may be granted or sold to participants in recognition of past services or for other valid consideration and may be issued in lieu of cash compensation due to such participant. The plan administrator may grant dividend equivalent rights to participants that entitle the recipient to receive credits for dividends that would have been paid if the recipient had held a specified number of shares of New GreenLight Common Stock.
Cash Awards
The plan administrator may grant cash-based awards under the New GreenLight Equity Plan to participants, subject to such vesting and other terms and conditions as the plan administrator may determine.
Payments by Participants
Participants in the New GreenLight Equity Plan are responsible for the payment of any federal, state, local or foreign taxes that New GreenLight or its subsidiaries are required by law to withhold upon the exercise of options or stock appreciation rights or vesting of other awards. The plan administrator may cause any tax withholding obligation of New GreenLight or its subsidiaries to be satisfied, in whole or in part, by the applicable entity withholding from shares of New GreenLight Common Stock to be issued pursuant to an award a number
134
of shares with an aggregate fair market value that would satisfy the withholding amount due. The plan administrator may also require any tax withholding obligation of New GreenLight or its subsidiaries to be satisfied, in whole or in part, by an arrangement whereby a certain number of shares issued pursuant to any award are immediately sold and proceeds from such sale are remitted to New GreenLight or its subsidiaries in an amount that would satisfy the withholding amount due.
Non-Transferability
of AwardsThe New GreenLight Equity Plan generally does not allow for the transfer or assignment of awards, other than by will or by the laws of descent and distribution or pursuant to a domestic relations order; however, the plan administrator may permit the transfer of nonstatutory stock options by option holders by gift to an immediate family member, to trusts for the benefit of family members, or to partnerships in which such family members are the only partners.
Merger or Change in Control
The New GreenLight Equity Plan provides that upon the effectiveness of a “change in control transaction,” as defined in the New GreenLight Equity Plan, an acquirer or successor entity may assume, continue or substitute for the outstanding awards under the New GreenLight Equity Plan. To the extent that awards granted under the New GreenLight Equity Plan are not assumed, continued or substituted by the successor entity, all awards granted under the New GreenLight Equity Plan shall terminate and, in such case (except as may be otherwise provided in the relevant award agreement), all stock options and stock appreciation rights with time-based vesting conditions or restrictions that are not vested and/or exercisable immediately prior to the effective time of the change in control transaction shall become fully vested and exercisable as of immediately prior to the effective time of the transaction, all other awards with time-based vesting conditions or restrictions shall become fully vested and nonforfeitable as of immediately prior to the effective time of the transaction, and all awards with conditions and restrictions relating to the attainment of performance goals may become vested and nonforfeitable in connection with the change in control transaction in the plan administrator’s discretion or to the extent specified in the relevant award agreement. In the event of such termination, individuals holding options and stock appreciation rights will, for each such award, either (a) receive a payment in cash or in kind for each share subject to such award that is exercisable in an amount equal to the per share value of the consideration payable to stockholders in the change in control transaction less the applicable per share exercise price (provided that, in the case of an option or stock appreciation right with an exercise price equal to or greater than the per share cash consideration payable to stockholders in the transaction, such option or stock appreciation right shall be cancelled for no consideration) or (b) be permitted to exercise such options and stock appreciation rights (to the extent exercisable) within a period of time prior to the transaction as specified by the plan administrator. The plan administrator shall also have the option (in its sole discretion) to make or provide for a payment, in cash or in kind, to participants holding other awards in an amount equal to the per share value of the consideration payable to stockholders in the change in control transaction multiplied by the number of vested shares under such awards.
Amendment or Termination
The plan administrator may establish subplans and modify exercise procedures and other terms and procedures in order to facilitate grants of awards subject to the laws and/or stock exchange rules of countries outside of the United States.
All awards will be subject to any New GreenLight clawback policy as set forth in such clawback policy or the applicable award agreement.
The New GreenLight Board may amend or discontinue the New GreenLight Equity Plan and the plan administrator may amend or cancel outstanding awards for purposes of satisfying changes in law or any other
135
lawful purpose, but no such action may materially and adversely affect rights under an award without the holder’s consent. Certain amendments to the New GreenLight Equity Plan will require the approval of New GreenLight’s stockholders.
New GreenLight 2022 Employee Stock Purchase Plan
The following is a summary of the principal features of the New GreenLight ESPP and its operation. This summary does not contain all of the terms and conditions of the New GreenLight ESPP and is qualified in its entirety by reference to the New GreenLight, which is filed as an exhibit to this annual report.
The New GreenLight ESPP has been adopted by the Board and approved by our stockholders. It is the intention of the Board that the New GreenLight ESPP qualify as an “employee stock purchase plan” under Section 423 of the Code. The Board believes that the adoption of the New GreenLight ESPP will benefit New GreenLight by providing employees with an opportunity to acquire shares of New GreenLight Common Stock and will help New GreenLight to attract, retain and motivate valued employees.
Purpose
The purpose of the New GreenLight ESPP is to provide eligible employees with an opportunity to purchase shares of New GreenLight Common Stock through accumulated contributions, which generally will be made through payroll deductions. The New GreenLight ESPP permits the administrator of the New GreenLight ESPP to grant purchase rights that qualify for preferential tax treatment under Section 423 of the Code. In addition, the New GreenLight ESPP authorizes the grant of purchase rights that do not qualify under Code Section 423 pursuant to rules, procedures or
sub-plans
adopted by the administrator that are designed to achieve desired tax or other objectives.Shares Available for Issuance
New GreenLight has initially reserved 2,000,000 shares of New GreenLight Common Stock for issuance under the New GreenLight ESPP. The New GreenLight ESPP provides that the number of shares of New GreenLight Common Stock reserved and available for issuance under such plan will automatically increase each January 1, beginning on January 1, 2023 and on each January 1 thereafter, by the least of 4,000,000 shares of New GreenLight Common Stock, 4% of the outstanding number of shares of New GreenLight Common Stock on the immediately preceding December 31, or such lesser amount as determined by the plan administrator. This limit is subject to adjustment in the event of a reorganization, recapitalization, reclassification, stock split, stock dividend, reverse stock split or other similar change in New GreenLight’s capitalization.
Administration
The New GreenLight ESPP will be administered by the compensation committee of the New GreenLight Board, the New GreenLight Board or another board committee pursuant to the terms of the ESPP. The plan administrator, which initially is the compensation committee of the New GreenLight Board, has full authority to make, administer and interpret such rules and regulations regarding the New GreenLight ESPP as it deems advisable.
Eligibility
Any employee of New GreenLight or one of its affiliates or subsidiaries that has been designated to participate in the New GreenLight ESPP is eligible to participate in the New GreenLight ESPP so long as the employee is customarily employed for at least 20 hours a week and more than five months in a calendar year. No person who owns or holds, or as a result of participation in the New GreenLight ESPP would own or hold, New GreenLight Common Stock or options to purchase New GreenLight Common Stock that together equal 5% or
136
more of the total combined voting power or value of all classes of capital stock of New GreenLight or any parent or subsidiary thereof is entitled to participate in the New GreenLight ESPP. No employee may be granted an option under the New GreenLight ESPP that permits the employee’s rights to purchase New GreenLight Common Stock to accrue at a rate of more than $25,000 (determined using the fair market value of the stock at the time such option is granted) in any calendar year.
Participation in the New GreenLight ESPP is limited to eligible employees who authorize payroll deductions equal to a whole percentage of base pay for allocation to the New GreenLight ESPP. Employees may authorize payroll deductions with a minimum of 1% of base pay and a maximum of 15% of base pay. As of December 31, 2021, approximately 281 employees would be eligible to participate in the New GreenLight ESPP. Once an employee becomes a participant in the New GreenLight ESPP, that employee will automatically participate in successive offering periods, as described below, until such time as that employee withdraws from the New GreenLight ESPP, becomes ineligible to participate in the New GreenLight ESPP, or his, her or their employment ceases.
Offering Periods and Purchase Periods
Unless otherwise determined by the plan administrator, each offering of New GreenLight Common Stock under the New GreenLight ESPP will be for a period of six months, which is referred to as an “offering period.” The first offering period under the New GreenLight ESPP will begin and end on such date or dates as determined by the plan administrator. Subsequent offerings under the New GreenLight ESPP will generally begin on the first business day occurring on or after each January 1 and July 1 and will end on the last business day occurring before the following July 1 and January 1, respectively. Shares are purchased on the last business day of each offering period, with that day being referred to as an “exercise date.” The plan administrator may establish different offering periods or exercise dates under the New GreenLight ESPP. The New GreenLight ESPP will include a component, or the “423 Component,” that is intended to qualify as an “employee stock purchase plan” under Code Section 423, and a component that does not comply with Code Section 423, or
the “Non-423 Component.”
For purposes of this summary, a reference to the New GreenLight ESPP generally will mean the terms and operations of the 423 Component.Except as may be permitted by the plan administrator in advance of an offering, a participant may not increase or decrease the amount of his, her or their payroll deductions during any offering period but may increase or decrease his, her or their payroll deduction with respect to the next offering period by filing a new enrollment form within the period beginning 15 business days before the first day of such offering period and ending on the day prior to the first day of such offering period. A participant may withdraw from an offering period at any time without affecting his, her or their eligibility to participate in future offering periods. If a participant withdraws from an offering period, that participant may not again participate in the same offering period, but may enroll in subsequent offering periods. An employee’s withdrawal will be effective as of the next business day following the date that the plan administrator receives the employee’s written notice of withdrawal under the New GreenLight ESPP.
Exercise of Purchase Right
On the exercise date of each offering period, the employee is deemed to have exercised the option, at the exercise price, for the lowest of (i) a number of shares of New GreenLight Common Stock determined by dividing such employee’s accumulated payroll deductions or contributions on such exercise date by the exercise price; (ii) the number of shares of New GreenLight Common Stock determined by dividing $25,000 by the fair market value of New GreenLight Common Stock on the first day of such offering period; or (iii) such lesser number as established by the plan administrator in advance of the offering. The exercise price is equal to the lesser of (i) 85% the fair market value per share of New GreenLight Common Stock on the first day of the offering period or (ii) 85% of the fair market value per share of New GreenLight Common Stock on the exercise date. The maximum number of shares of New GreenLight Common Stock that may be issued to any employee
137
under the New GreenLight ESPP in a calendar year is a number of shares of New GreenLight Common Stock determined by dividing $25,000 by the fair market value of New GreenLight Common Stock, valued at the start of the offering period, or such other lesser number of shares as determined by the plan administrator from time to time.
Termination of Participation
In general, if an employee is no longer a participant on an exercise date, the employee’s option will be automatically terminated, and the amount of the employee’s accumulated payroll deductions will be refunded.
Non-Transferability
A participant will not be permitted to transfer rights granted under the New GreenLight ESPP other than by will or the laws of descent and distribution, and such rights are generally exercisable during the lifetime of the participant only by the participant.
Merger or Change in Control
In the case of and subject to the consummation of a “change in control,” the plan administrator, in its discretion, and on such terms and conditions as it deems appropriate, may take any one or more of the following actions under the New GreenLight ESPP or with respect to any right under the New GreenLight ESPP or to facilitate such transactions or events: (a) provide for either (i) termination of any outstanding option in exchange for an amount of cash, if any, equal to the amount that would have been obtained upon the exercise of such option had such option been currently exercisable or (ii) the replacement of such outstanding option with other options or property selected by the plan administrator in its sole discretion; (b) provide that the outstanding options under the New GreenLight ESPP shall be assumed by the successor or survivor corporation, or a parent or subsidiary thereof, or shall be substituted for similar options covering the stock of the successor or survivor corporation, or a parent or subsidiary thereof, with appropriate adjustments as to the number and kind of securities and prices; (c) make adjustments in the number and type of shares of New GreenLight Common Stock (or other securities or property) subject to outstanding options under the New GreenLight ESPP and/or in the terms and conditions of outstanding options and options that may be granted in the future; (d) provide that the offering with respect to which an option relates will be shortened by setting a new exercise date on which such offering will end; and (e) provide that all outstanding options shall terminate without being exercised and all amounts in the accounts of participants shall be promptly refunded.
Amendment; Termination
The New GreenLight ESPP will automatically terminate on the tenth anniversary of the effective date of the New GreenLight ESPP. The New GreenLight Board may, in its discretion, at any time, terminate or amend the New GreenLight ESPP. However, without the approval within 12 months of such New GreenLight Board action by the stockholders, no amendment shall be made to the New GreenLight ESPP increasing the number of shares specifically approved to comply with the requirements of Section 423(b) of the Code or any other changes to the components of the New GreenLight ESPP intended to comply with the requirements of Section 423(b) of the Code that would require stockholder approval in order for the New GreenLight ESPP, as amended, to qualify as an “employee stock purchase plan” under Section 423(b) of the Code.
GreenLight Director Compensation
Unless the context otherwise requires, any reference in this section of this Annual Report to “GreenLight,” “we,” “us” or “our” refer to GreenLight and its consolidated subsidiaries prior to the consummation of the Business Combination and to New GreenLight and its consolidated subsidiaries following the Business Combination.
138
GreenLight currently has no formal policy underincurred in connection with attending board and committee meetings or performing other services in their capacities
which non-employee directors
receive compensation for their service on the GreenLight Board or its committees.Certain non-employee directors
receive cash fees for their service as a director of GreenLight. GreenLight’s policy is toreimburse non-employee directors
for reasonable andnecessary out-of-pocket expenses
as non-employee directors,
and GreenLight occasionally grants stock optionsto non-employee directors.
Director Compensation Table
The following table provides information regarding the compensation of each person serving as a director of GreenLight for 2021, other than Dr. Zarur, our President and Chief Executive Officer. Dr. Zarur does not receive any compensation for service in his capacity as a director. His compensation as a named executive officer is set forth above in “—Executive Compensation.”
Name | Fees earned or paid in cash ($) | Total ($) | ||||||
Dr. Charles Cooney | 75,000 | 75,000 | ||||||
Jason Dinges | — | — | ||||||
Mike Liang | — | — | ||||||
Dr. Ganesh Kishore | — | — | ||||||
Eric O’Brien | — | — | ||||||
Dr. Martha Schlicher (1) | 50,000 | 50,000 | ||||||
| (1) | At December 31, 2021, Dr. Schlicher held 4,213 shares of restricted stock acquired upon the early exercise of a stock option granted prior to 2021. These shares vest in two parts: (a) 213 shares vest in 1 monthly installment of 104 shares and a final installment of 109 shares and (b) 4,000 shares vest in two equal annual installments, the first of which shall vest on June 24, 2022. |
Each of David Brewster, Daniel Coyne, Jennifer E. Pardi, Deval Patrick and Dean Seavers served as a director of ENVI during 2021. At the closing of ENVI’s initial public offering in January 2021, ENVI issued 50,000 insider warrants to each of Mr. Brewster, Gov. Patrick and Mr. Seavers in connection with services to be rendered by ENVI’s management team in connection with the initial public offering and ENVI’s business combination activities. Such warrants are identical to the Private Placement Warrants, including as to exercise price, exercisability and exercise period. The warrants issued to each director had an estimated grant date fair value of $59,500. For more information regarding these warrants, see Exhibit 4.3, “” Except for these warrants, none of ENVI’s directors received any cash compensation or other
Description of Capital Stock—Warrants—Private Placement Warrants.
non-cash
compensation for their service as directors.New GreenLight Director Compensation
The New GreenLight Board expects to review director compensation periodically to ensure that director compensation remains competitive so that New GreenLight will be able to recruit and retain qualified directors. In 2020, the Talent and Compensation Committee of the GreenLight Board retained Pay Governance LLC, a third-party compensation consultant, to provide the Talent and Compensation Committee and the GreenLight Board with an analysis of publicly available market data regarding practices and compensation levels at comparable companies and assistance in determining compensation to be provided to New GreenLight
non-employee
directors. Based on the discussions with and assistance from the compensation consultant, it is expected that, following the Business Combination, New GreenLight will provide the compensation described below to certain of the NewGreenLight non-employee directors.
139
Cash Compensation
New GreenLight expects to recommend the following cash compensation to the New GreenLight compensation committee for approval to New
GreenLight non-employee directors:
| • | $50,000 per year for service as a non-employee director (other than the chair); |
| • | $75,000 per year for service as non-employee chair of the New GreenLight Board; |
| • | $15,000 per year for service as chair of the New GreenLight audit committee; |
| • | $7,500 per year for service as a member of the New GreenLight audit committee (other than the chair); |
| • | $10,000 per year for service as chair of the New GreenLight talent and compensation committee; |
| • | $5,000 per year for service as a member of the New GreenLight talent and compensation committee (other than the chair); |
| • | $8,000 per year for service as chair of the New GreenLight nominating and corporate governance committee; and |
| • | $4,000 per year for service as a member of the New GreenLight nominating and corporate governance committee (other than the chair). |
A non-employee director
who serves asthe non-employee chair
of the New GreenLight Board will receive the annual retainer fee for that role, which includes the annual retainer fee for service asa non-employee director.
Each non-employee director
who serves as a committee chair of the New GreenLight Board will receive the cash retainer fee for service as the chair of the committee but not the cash retainer fee for service as a member of that committee. These fees to NewGreenLight non-employee directors
are expected to be paid quarterly in arrears on a prorated basis. The above-listed fees for service as a chair or member of any committee are payable in addition tothe non-employee director
retainer. It is expected that New GreenLight will also reimburseits non-employee directors
for reasonable travel expenses to attend meetings of the New GreenLight Board and its committees.Equity Compensation
Initial Award
a non-employee director,
on or after the date that the person first becomesa non-employee director,
an initial award of stock options to purchase shares of New GreenLight Common Stock (the “Initial Award
”). The value of the Initial Award has not yet been determined. Each Initial Award is expected to vest pursuant to its vesting schedule, subject to continued services to New GreenLight through the applicable vesting dates.Annual Award
each non-employee director,
on or after the date of each annual meeting of New GreenLight stockholders (an “Annual Meeting
”), an award of stock options to purchase shares of New GreenLight Common Stock (the “Annual Award
”). The value of the Annual Award has not yet been determined. Each Annual Award is expected to vest pursuant to its vesting schedule, subject to continued services to New GreenLight through the applicable vesting dates.Minimum Ownership Requirements
Other Award Terms
140
Director Compensation Limits
any non-employee director
in any calendar year shall not exceed: (i) $500,000 in the first calendar year an individual becomes a non- employee director and (ii) $375,000 in any other calendar year. This limitation will be determined without regard to amounts paid toa non-employee director
(including retirement benefits and severance payments) in respect of any services provided in any capacity (including employee or consultant) other than asa non-employee director.
The Board may make exceptions to this limit fora non-executive chair
of the Board with the approval of a majority of the disinterested directors.ITEM 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
Principal Stockholders
The following table provides information regarding the beneficial ownership of New GreenLight Common Stock as of March 15, 2022 by:
| • | each person known by New GreenLight to be the beneficial owner of more than 5% of New GreenLight Common Stock; |
| • | each director and named executive officer of New GreenLight; and |
| • | all directors and executive officers of New GreenLight as a group. |
Beneficial ownership is determined according to the rules of the Commission, which generally provide that a person has beneficial ownership of a security if the person possesses sole or shared voting or investment power over that security, including options and warrants that are currently exercisable or that will become exercisable within 60 days after March 15, 2022. Unless otherwise indicated, New GreenLight believes that all persons named in the table below have sole voting and investment power with respect to the voting securities beneficially owned by them.
The beneficial ownership of New GreenLight Common Stock is based on 122,839,613 shares of New GreenLight Common Stock issued and outstanding as of March 15, 2022:
Name of Beneficial Owner | Number | Percentage | ||||||
Five Percent or Greater Holders | ||||||||
Builders Vision, LLC (1) | 15,843,021 | 12.9 | % | |||||
Morningside Venture Partners (2) | 13,857,931 | 11.3 | % | |||||
Kodiak Venture Partners (3) | 9,809,892 | 8.0 | % | |||||
Fall Line Endurance Fund, LP (4) | 8,901,814 | 7.2 | % | |||||
Cormorant Asset Management, LP (5) | 6,710,540 | 5.5 | % | |||||
Directors and Named Executive Officers | ||||||||
Matthew Walker (1) | 15,843,021 | 12.9 | % | |||||
Eric O’Brien (4) | 8,901,812 | 7.2 | % | |||||
Ganesh Kishore (6) | 5,818,575 | 4.7 | % | |||||
Dr. Andrey Zarur (7) | 3,787,853 | 3.1 | % | |||||
Carole Cobb (8) | 1,408,216 | 1.1 | % | |||||
Susan E. Keefe (8) | 485,310 | * | ||||||
Charles Cooney | 305,314 | * | ||||||
Martha Schlicher (9) | 109,218 | * | ||||||
Jennifer Pardi | — | — | ||||||
All directors and executive officers as a group (14 individuals) | 37,269,880 | 30.3 | % | |||||
| * | Indicates beneficial ownership less than 1%. |
141
| (1) | Includes (a) 3,673,694 shares held by S2G Builders Food & Agriculture Fund III, LP (“ Fund III ”); (b) 2,087,043 shares held by S2G Ventures Fund I, L.P. (“Fund I ”); and (c) 8,582,284 shares held by S2G Ventures Fund II, L.P. (“Fund II ” and, together with Fund I and Fund III, the “S2G Funds ”). Builders Vision, LLC is the Manager of Funds I and II, and the General Partner of Fund III, and has power to vote or direct the voting of shares held by the S2G Funds. The General Partners of Fund I and Fund II are S2G Ventures, LLC and S2G Ventures II, LLC, respectively. Mr. Walker, a director of New GreenLight and a former director of GreenLight, is a Managing Director of Builders Vision, LLC, the impact platform founded by Lukas T. Walton, which includes S2G Ventures. By virtue of the foregoing, S2G Ventures, LLC, S2G Ventures II, LLC, and Mr. Walton may be deemed to indirectly beneficially own (as defined in Rule13d-3 of the Exchange Act) the shares of New GreenLight Common Stock held by the S2G Funds. Mr. Walker and Mr. Walton each disclaims beneficial ownership of these shares of New GreenLight Common Stock except to the extent of any pecuniary interest therein. The business address for Builders Visions, LLC is P.O. Box 1860, Bentonville, Arkansas 72712. |
| (2) | Represents (a) 12,857,931 shares held by Morningside Venture Investments Limited, and (b) 1,000,000 shares held by MVIL, LLC (together with Morningside Venture Investments Limited, “ Morningside de Galles, 3-5 Avenue des Citronniers, MC 98000, Monaco. |
| (3) | Includes (a) 9,573,157 shares held by Kodiak Venture Partners III, L.P., and (b) 236,738 shares held by Kodiak III Entrepreneurs Fund, L.P. (together with Kodiak Venture Partners III, L.P. “ Kodiak Kodiak Ventures ”) is the General Partner for Kodiak, Kodiak Venture Management (GP), LLC is the General Partner for Kodiak Ventures and Kodiak Ventures Management Company, Inc. is the Member of Kodiak Ventures Management (GP), LLC (“Kodiak Ventures Management ”). Mr. David Furneaux is the Chief Executive Officer of Kodiak Ventures Management Company, Inc. Each therefore has the power to vote, or direct the voting of, the shares of New GreenLight Common Stock held by Kodiak. By virtue of the foregoing, each of Kodiak Ventures Management and Mr. Furneaux may be deemed to indirectly beneficially own (as that term is defined in Rule13d-3 of the Exchange Act) the New GreenLight Common Stock held by Kodiak. Mr. Furneaux disclaims beneficial ownership of these shares of New GreenLight Common Stock except to the extent of any pecuniary interest therein. The business address for Kodiak, Kodiak Ventures Management and Mr. Furneaux is 11 Peter Grover Road, Bethel, Maine 04217. |
| (4) | Represents shares held by Fall Line Endurance Fund, LP (“ Fall Line ”). Mr. Eric O’Brien, a director of New GreenLight, is theco-founder and Managing Director of Fall Line and has the power to vote, or to direct the voting of, the shares of New GreenLight Common Stock held by Fall Line. By virtue of the foregoing, Mr. O’Brien may be deemed to indirectly beneficially own (as that term is defined in Rule13d-3 of the Exchange Act) the shares of New GreenLight Common Stock held by Fall Line. Mr. O’Brien disclaims beneficial ownership of these shares of New GreenLight Common Stock except to the extent of any pecuniary interest therein. The business address of Fall Line and Mr. O’Brien is 119 South B Street, Suite B, San Mateo, CA 94401. |
| (5) | Includes (a) 2,272,901 shares of New GreenLight Common Stock held by Cormorant Global Healthcare Master Fund, LP (“ Master Fund ”), and (b) 4,437,639 shares of New GreenLight Common Stock held by Cormorant Private Healthcare Fund II, LP (“Fund II ”). Cormorant Global Healthcare GP, LLC serves as the General Partner of Master Fund and Cormorant Private Healthcare GP II, LLC serves as the General Partner of Fund II. Cormorant Asset Management, LP serves as the investment manager to Master Fund and Fund II. Bihua Chen serves as the Managing Member of Cormorant Global Healthcare GP, LLC, Cormorant Private Healthcare GP II, LLC and the general partner of Cormorant Asset Management, LP (together with Master Fund and Fund II, the “Cormorant Entities ”). By virtue of the foregoing, each of Bihua Chen and the Cormorant Entities may be deemed to indirectly beneficially own (as that term is defined in Rule13d-3 of the Exchange Act) the shares held by each of the relevant Cormorant Entities. Each of Bihua Chen and the Cormorant Entities disclaims beneficial ownership of such shares except to the extent of her or its pecuniary |
142
| interest therein. The business address of each of Bihua Chen and the Cormorant Entities is 200 Clarendon St., 52nd Floor, Boston, Massachusetts. |
| (6) | Represents shares held by MLS Capital Fund II, L.P. (“ MLS ”). Mr. Kishore, a director of New GreenLight, is aCo-Manager of MLSCF II (GP) (Labuan), LLP, the General Partner of MLS, and has the power to vote, or to direct the voting of, the shares held by MLS. By virtue of the foregoing, Mr. Kishore may be deemed to indirectly beneficially own (as that term is defined in Rule13d-3 of the Exchange Act) the shares held by MLS. Mr. Kishore disclaims beneficial ownership of these shares except to the extent of any pecuniary interest therein. The business address of MLS and Mr. Kishore is c/o Spruce Capital Partners LLC, 660 4th Street, #295, San Francisco, California 94107. |
| (7) | Includes (a) 896,058 shares and (b) 2,891,795 shares subject to options exercisable within 60 days after March 15, 2022. |
| (8) | Represents shares subject to options exercisable within 60 days after March 15, 2022. |
| (9) | Includes 4,000 shares subject to vesting as of March 15, 2022, as follows: 2,000 shares shall vest on June 24, 2022 and 2,000 shares shall vest on June 24, 2023. |
Securities Authorized for Issuance under Equity Compensation Plans
ENVI
The following table provides information as of December 31, 2021 regarding shares authorized for issuance under ENVI’s equity compensation plans, including individual compensation arrangements. As of that date, ENVI had no equity compensation plans or individual compensation arrangements, other than an aggregate of 150,000 Private Placement Warrants issued in January 2021 to ENVI’s three independent directors for services to be rendered in connection with its initial public offering and business combination activities (the “”).
ENVI Director Warrants
Plan category | Number of securities to be issued upon exercise of outstanding options, warrants and rights (a) | Weighted-average exercise price of outstanding options, warrants and rights (b) | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) (c) | |||||||||
Equity compensation plans approved by security holders | — | — | — | |||||||||
Equity compensation plans not approved by security holders | 150,000 | $ | 11.50 | — | ||||||||
Total | 150,000 | $ | 11.50 | — | ||||||||
New GreenLight
On February 1, 2022, the ENVI stockholders approved the proposal to adopt the New GreenLight Equity Plan, pursuant to which an aggregate of 31,750,000 shares of New GreenLight Common Stock were authorized for issuance, including pursuant to the Rollover Options, and the proposal to adopt the New GreenLight ESPP, pursuant to which an aggregate of 2,000,000 shares of common stock were authorized for issuance.
Each of the New GreenLight Equity Plan and the New GreenLight ESPP provides that the number of shares reserved and available for issuance under such plan will automatically increase each January 1, beginning on January 1, 2023, by 4% of the outstanding number of shares of New GreenLight Common Stock on the immediately preceding December 31, or such lesser amount as determined by the New GreenLight Board.
143
The following table provides information as of immediately after the closing of the Business Combination on February 2, 2022 regarding shares authorized for issuance under the New GreenLight Equity Plan, the New GreenLight ESPP and the ENVI Director Warrants. There were no other equity compensation plans or individual compensation arrangements outstanding at that time. The following table does not reflect grants, exercises, forfeitures and other activity after February 2, 2022.
Plan category | Number of securities to be issued upon exercise of outstanding options, warrants and rights (a) | Weighted-average exercise price of outstanding options, warrants and rights (b) | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) (c) | |||||||||
Equity compensation plans approved by security holders | 17,933,794 | (1) | $ | 1.16 | 13,816,206 | (2) | ||||||
Equity compensation plans not approved by security holders | 150,000 | $ | 11.50 | — | ||||||||
Total | 18,083,794 | $ | 1.24 | 13,816,206 | (2) | |||||||
| (1) | Does not include 4,109 shares of restricted stock granted under the Equity Plan which were not vested as of February 2, 2022 and therefore subject to forfeiture. |
| (2) | Includes 2,000,000 shares of common stock reserved for issuance under the New GreenLight ESPP. |
ITEM 13. | Certain Relationships and Related Transactions and Director Independence |
Described below are any transactions occurring since January 1, 2019 or, in the case of ENVI, July 2, 2020 (its date of formation) and any currently proposed transactions to which either ENVI (now New GreenLight) or GreenLight was a party and in which:
| • | the amounts involved exceeded or will exceed the lesser of $120,000 or 1% of the average of ENVI’s or GreenLight’s total assets, as applicable, at year-end for the last two completed fiscal years; and |
| • | a director, executive officer, holder of more than 5% of the outstanding capital stock of ENVI or GreenLight, or any member of such person’s immediate family, had or will have a direct or indirect material interest. |
Certain Relationships and Related Person Transactions—ENVI
Class B Common Stock
In connection with ENVI’s initial formation in July 2020, the Sponsor and HB Strategies were issued all of ENVI’s outstanding equity. The Sponsor purchased 2,156,250 shares of ENVI Class B Common Stock in August 2020, resulting in the Sponsor directly owning such shares of ENVI Class B Common Stock. The remaining 5,031,250 shares of ENVI Class B Common Stock were purchased by HB Strategies in September 2020. In December 2020, the Sponsor and HB Strategies returned to ENVI, at no cost, 862,500 and 2,443,750 shares of ENVI Class B Common Stock, respectively, and ENVI issued 143,750 shares of ENVI Class B Common Stock to each of Gov. Patrick, Messrs. Brewster and Seavers, our independent directors, resulting in an aggregate of 4,312,500 shares of ENVI Class B Common Stock outstanding and held by the Initial Stockholders. On January 13, 2021, ENVI effected a stock dividend of 1.2 shares for each share of ENVI Class B Common Stock outstanding, resulting in the Initial Stockholders holding an aggregate of 5,175,000 shares of ENVI Class B Common Stock (up to 675,000 shares of which were subject to forfeiture depending on the extent to which the underwriters’ over-allotment option was exercised). The number of shares of ENVI Class B Common Stock outstanding was determined based on the expectation that such shares would represent 20% of the outstanding shares after the initial public offering.
144
The Initial Stockholders agreed, subject to limited exceptions, not to transfer, assign or sell any of their shares of ENVI Class B Common Stock until the earlier to occur of: (A) six months after the completion of a business combination and (B) subsequent to a business combination, (x) if the last sale price of the ENVI Class A Common Stock equals or exceeds $12.00 per share (as adjusted for stock splits, stock capitalizations, reorganizations, recapitalizations and the like) for any 20 trading days within
any 30-trading day
period commencing at least 60 days after a Business Combination, or (y) the date on which ENVI completes a liquidation, merger, capital stock exchange or other similar transaction that results in all of the public stockholders having the right to exchange their shares of common stock for cash, securities or other property.Private Placement Warrants
Simultaneously with the closing of the initial public offering, HB Strategies purchased an aggregate of 2,000,000 Private Placement Warrants (the “
Private Placement Warrants
”) at a price of $1.00 per Private Placement Warrant in a private placement, generating gross proceeds of $2.0 million. At that same time, ENVI also issued to the Sponsor 600,000 warrants pursuant to a Warrant Subscription Agreement dated December 21, 2020 (the “Sponsor Warrants
”), and issued each of ENVI’s three independent directors 50,000 warrants pursuant to certain warrant grant agreements dated December 21, 2020 (the “Director Warrants
”). The Private Placement Warrants, Sponsor Warrants and Director Warrants are substantially identical to the Warrants included in the units sold as part of the units in ENVI’s initial public offering. No underwriting discounts or commissions were paid with respect to such sale. The issuances of the Private Placement Warrants, Sponsor Warrants and Director Warrants were made pursuant to the exemption from registration contained in Section 4(a)(2) of the Securities Act.At the closing of the Business Combination, pursuant to the terms of the Sponsor Letter Agreement, HB Strategies and the Sponsor forfeited an aggregate of 687,500 Private Placement Warrants and Sponsor Warrants.
The private placement shares are subject to
the lock-up period
described in the Investor Rights Agreement that was executed by the Initial Stockholders in connection with the execution of the Business Combination Agreement.Related Party Note
On September 4, 2020, HB Strategies issued the Promissory Note, pursuant to which ENVI was entitled to borrow up to an aggregate principal amount of $300,000. The Promissory Note
was non-interest bearing
and payable on the earlier of (i) March 31, 2021 or (ii) the consummation of the initial public offering. The Promissory Note was repaid at the closing of the initial public offering on January 19, 2021. On August 9, 2021, ENVI issued a promissory note to HB Strategies in the aggregate principal amount of $500,000, which note wasnon-interest
bearing and payable on the earlier of (i) January 19, 2022 or (ii) the consummation of an initial business combination. In connection with the Closing of the Business Combination, this note was forgiven in full.Related Party Loans
In order to finance transaction costs in connection with a business combination, the Sponsor, members of ENVI management or any of their respective affiliates or other third parties were entitled to, but not obligated to, loan ENVI funds as may be required (“
Working Capital Loans
”), which were to be repaid only upon the consummation of a business combination. If ENVI does not consummate a business combination, ENVI may use a portion of any funds held outside the Trust Account to repay the Working Capital Loans; however, no proceeds from the Trust Account may be used for such repayment. Up to $1,500,000 of the Working Capital Loans were entitled to be converted into units identical to the ENVI Units at a price of $10.00 per unit at the option of the holder. As of the Closing of the Business Combination, there were no Working Capital Loans outstanding.145
Business Combination Marketing Agreement
On January 13, 2021 ENVI entered into the Business Combination Marketing Agreement with Canaccord, an affiliate of the Sponsor, pursuant to which ENVI engaged Canaccord as an advisor in connection with a business combination, to assist ENVI in arranging meetings with its stockholders to discuss the potential business combination and the target business’ attributes, introduce ENVI to potential investors that may be interested in purchasing ENVI securities, assist in obtaining stockholder approval for the business combination and assist with the preparation of ENVI press releases and public filings in connection with the business combination. For such services, ENVI agreed to will pay Canaccord upon the consummation of a business combination, including the Business Combination, a cash fee in an amount equal to 3.76% of the gross proceeds of the initial public offering (or $7,783,200) (exclusive of any applicable finders’ fees which might become payable). Pursuant to the terms of the Business Combination Marketing Agreement, no fee would have been if ENVI did not complete a business combination. At the Closing of the Business Combination, the parties agreed to reduce the cash fee to $5.8 million, and such amount was paid.
Investor Rights Agreement
Concurrently with the execution of the Business Combination Agreement, ENVI, the Initial Stockholders and certain stockholders of GreenLight entered into the Investor Rights Agreement pursuant to which, among other things, the Initial Stockholders and such stockholders of GreenLight agreed not to effect any sale or distribution of any equity securities of ENVI during
the lock-up period
described therein and were granted certain registration rights, in each case subject to, and conditioned upon and effective as of, the effective time of the Merger. The Investor Rights Agreement amended and restated ENVI’s previous registration rights agreement with its Initial Stockholders.For additional information, see Exhibit 4.3, “”
Description of Capital Stock—Registration Rights—Investor Rights Agreement.
Sponsor Letter Agreement
Concurrently with the execution of the Business Combination Agreement, the Initial Stockholders and GreenLight entered into the Sponsor Letter Agreement, pursuant to which the Initial Stockholders agreed to, among other things, (i) vote all of their founder shares in favor of, and consent to, the Business Combination Agreement and the transactions contemplated thereby (including the Merger), (ii) waive any adjustment to the conversion ratio set forth in the Former Organizational Documents or any other anti-dilution or similar protection with respect to the Class B Shares, whether resulting from the transactions contemplated by the Business Combination Agreement or otherwise, and (iii) be bound by certain other covenants and agreements related to the Business Combination including an agreement to deal exclusively with GreenLight and restrictions on transfers with respect to his, her or its founder shares prior to the Closing. However, shares of ENVI Class A Common Stock owned by HB Strategies are not subject to certain of the transfer restrictions under the Sponsor Letter Agreement. In addition, HB Strategies and the Sponsor agreed that, if more than 25% of the shares of ENVI Class A Common Stock were redeemed pursuant to the Former Charter, then they would forfeit 25% of the private placement and private placement-equivalent warrants immediately before the closing of the Business Combination. Because redemptions exceed the stated threshold, the requisite number of warrants was forfeited upon the Closing of the Business Combination.
Certain Relationships and Related Person Transactions—GreenLight Biosciences
GreenLight Convertible Note Financing
From April 2020 through May 2020, GreenLight Pandemic Response, Inc. (“
GPR
”), a wholly owned subsidiary of GreenLight, sold GreenLight Convertible Notes (the “GreenLight Convertible Notes
”) with an aggregate principal amount of $16.8 million for an aggregate purchase price of $16.8 million (the “GreenLight
146
Convertible Note Financing
”). The GreenLight Convertible Notes are unsecured, mature between April 2022 and May 2022 and bear simple interest at the rate of 5% per annum. GreenLight unconditionally guaranteed both payment and performance of the GreenLight Convertible Notes. At the time of issuance, the GreenLight Convertible Notes were convertible at the election of the holder into either (a) shares of GreenLight’s then-anticipated Series D Preferred Stock (the “Share Conversion Right
”) or (b) a right to receive a royalty payment based on revenue generated from GPR’s proposedCOVID-19
business.The participants in the GreenLight Convertible Note Financing included persons affiliated with members of GreenLight’s board of directors and persons that held more than 5% of GreenLight’s outstanding capital stock at the time of the issuance of the GreenLight Convertible Notes. The following table summarizes purchases of GreenLight Convertible Notes by such related persons:
Name | GreenLight Convertible Note Principal Amount | Total Purchase Price | ||||||
S2G Ventures Fund II, L.P. (1) | $ | 3,000,000 | $ | 3,000,000 | ||||
Fall Line Endurance Fund, LP (2) | $ | 2,000,000 | $ | 2,000,000 | ||||
Baird Venture Partners V Limited Partnership (3) | $ | 1,662,130 | $ | 1,662,130 | ||||
BVP V Affiliates Fund Limited Partnership (3) | $ | 174,006 | $ | 174,006 | ||||
BVP Special Affiliates Limited Partnership (3) | $ | 163,864 | $ | 163,864 | ||||
| (1) | Matthew Walker was a member of the GreenLight board of directors at the time of the issuance of the GreenLight Convertible Notes and continuously served on the GreenLight board of directors until the Closing of the Business Combination. S2G Ventures Fund II, L.P. (“ S2G II ”) and its affiliated funds held more than 5% of GreenLight’s outstanding capital stock at the time of issuance of the GreenLight Convertible Notes to S2G II and continuously held more than 5% of GreenLight’s outstanding capital stock at all times since the issuance of the GreenLight Convertible Notes until the Closing of the Business Combination. Mr. Walker became a New GreenLight director upon the Closing of the Business Combination. |
| (2) | Eric O’Brien was a member of the GreenLight board of directors at the time of the issuance of the GreenLight Convertible Notes and continuously served on the GreenLight board of directors since that time until the Closing of the Business Combination. During this period, he has been an affiliate of Fall Line Endurance Fund L.P. (“ Fall Line ”). Fall Line held more than 5% of GreenLight’s outstanding capital stock at the time of issuance of the GreenLight Convertible Notes to Fall Line and continuously held more than 5% of GreenLight’s outstanding capital stock at all times since the issuance of the GreenLight Convertible Notes until the Closing of the Business Combination. Mr. O’Brien became a New GreenLight director upon the Closing of the Business Combination. |
| (3) | Michael Liang was a member of the GreenLight board of directors at the time of the issuance of the GreenLight Convertible Notes and continuously served on the GreenLight board of directors since that time until the Closing of the Business Combination. During this period, he has been an affiliate of each of Baird Venture Partners V Limited Partnership, BVP V Affiliates Fund Limited Partnership and BVP Special Affiliates Limited Partnership (all such funds collectively, “ Baird ”). Baird held less than 5% of GreenLight’s outstanding capital stock at the time of GreenLight Convertible Note Financing. |
In connection with the issuance of the GreenLight Convertible Notes, GreenLight entered into side letters with each of S2G II, Fall Line and Baird in which GreenLight and such investor agreed, among other things, that, if the then-anticipated GreenLight Series D Preferred Stock Financing were to be led by an existing investor in GreenLight, the conversion rate of the GreenLight Convertible Notes would be reduced. This condition was not satisfied.
147
In August 2021, with the consent of the holders of a majority of the aggregate outstanding principal amount of GreenLight Convertible Notes, we amended and restated such notes to eliminate the right to receive a royalty and to make GreenLight the direct and sole obligor on such notes.
At the Closing, the entire outstanding amount of principal and unpaid interest under the GreenLight Convertible Notes converted in accordance with their terms and the terms of the Business Combination Agreement, into an aggregate of 6,719,110 shares of New GreenLight Common Stock, for an effective purchase price of $1.8118 per share.
GreenLight Series D Preferred Stock Financing
In June and July 2020, GreenLight sold an aggregate of 60,184,332 shares of GreenLightSeries D Preferred Stock at a purchase price of $1.8118 per share, or an aggregate purchase price of $109.0 million (the “
GreenLight Series D Preferred Stock Financing
”).The participants in the GreenLight Series D Preferred Stock Financing included persons affiliated with members of GreenLight’s board of directors and persons that held more than 5% of GreenLight’s outstanding capital stock at the time of the closing of the GreenLight Series D Preferred Stock Financing. The following table summarizes purchases of shares of GreenLight Series D Preferred Stock from GreenLight by such related persons:
Name | Number of Shares | Total Purchase Price | ||||||
Morningside Venture Investments Limited (1) | 19,317,805 | $ | 34,999,999 | |||||
S2G Builders Food & Agriculture Fund III, LP (2) | 5,519,372 | $ | 9,999,998 | |||||
S2G Ventures Fund II, L.P. (2) | 3,863,561 | $ | 7,000,000 | |||||
Fall Line Endurance Fund, LP (3) | 3,311,623 | $ | 5,999,999 | |||||
Baird Venture Partners V Limited Partnership (4) | 2,064,131 | $ | 3,739,793 | |||||
BVP Special Affiliates Limited Partnership (4) | 216,090 | $ | 391,512 | |||||
BVP V Affiliates Fund Limited Partnership (4) | 203,495 | $ | 368,692 | |||||
Series Greenlight 2, A Separate Series of BlueIO Growth LLC (5) | 1,421,238 | $ | 2,574,999 | |||||
MLS Capital Fund II, L.P. (6) | 1,103,874 | $ | 1,999,999 | |||||
| (1) | Jason Dinges was a member of the GreenLight board of directors at the time of the GreenLight Series D Preferred Stock Financing. Mr. Dinges joined the GreenLight board of directors at the time of the closing of the GreenLight Series D Preferred Stock Financing and continuously served on the GreenLight board of directors since that time until the Closing of the Business Combination. Morningside acquired more than 5% of GreenLight’s outstanding capital stock in the GreenLight Series D Preferred Stock Financing. |
| (2) | Matthew Walker was a member of the GreenLight board of directors at the time of the GreenLight Series D Preferred Stock Financing and continuously served on the GreenLight board of directors since that time until the Closing of the Business Combination. S2G Ventures held more than 5% of GreenLight’s outstanding capital stock at the time of the GreenLight Series D Preferred Stock Financing and continuously held more than 5% of GreenLight’s outstanding capital stock at all times since the GreenLight Series D Preferred Stock Financing until the Closing of the Business Combination. Mr. Walker became a New GreenLight director upon the Closing of the Business Combination. |
| (3) | Eric O’Brien was a member of the GreenLight board of directors at the time of the GreenLight Series D Preferred Stock Financing and continuously served on the GreenLight board of directors since that time until the Closing of the Business Combination. During this period, he has been an affiliate of Fall Line. Fall Line held more than 5% of GreenLight’s outstanding capital stock at the time of the GreenLight Series D Preferred Stock Financing and continuously held more than 5% of GreenLight’s outstanding capital stock at all times since the GreenLight Series D Preferred Stock Financing until the Closing of the Business Combination. Mr. O’Brien became a New GreenLight director upon the Closing of the Business Combination. |
148
| (4) | Michael Liang was a member of the GreenLight board of directors at the time of the GreenLight Series D Preferred Stock Financing and continuously served on the GreenLight board of directors since that time until the Closing of the Business Combination. During this period, he has been an affiliate of Baird. Baird held less than 5% of GreenLight’s outstanding capital stock at the time of GreenLight Series D Preferred Stock Financing. |
| (5) | David Furneaux was a member of the GreenLight board of directors at the time of the GreenLight Series D Preferred Stock Financing. Mr. Furneaux joined the board of directors in July 2013 and served on the GreenLight board of directors until the initial closing of the GreenLight Series D Preferred Stock Financing. During this period, he was an affiliate of Series Greenlight 2, A Separate Series of BlueIO Growth LLC (“ Series Greenlight 2 ”). Series Greenlight 2 held less than 5% of GreenLight’s outstanding capital stock at the time of GreenLight’s Series D Preferred Stock Financing. |
| (6) | Ganesh Kishore was a member of the GreenLight board of directors at the time of the GreenLight Series D Preferred Stock Financing and continuously served on the GreenLight board of directors since that time until the Closing of the Business Combination. During this period, he has been an affiliate of MLS Capital Fund II, L.P. (“ MLS ”). MLS held more than 5% of GreenLight’s outstanding capital stock at the time of the GreenLight Series D Preferred Stock Financing and continuously held more than 5% of GreenLight’s outstanding capital stock at all times since the GreenLight Series D Preferred Stock Financing until the Closing of the Business Combination. Mr. Kishore became a New GreenLight director upon the Closing of the Business Combination. |
GreenLight Series C Preferred Stock Financing
From December 2018 to June 2019, GreenLight sold an aggregate of 35,092,183 shares of its Series C Preferred Stock at a purchase price of $1.59 per share, or an aggregate purchase price of $55.7 million (the “
GreenLight Series C Preferred Stock Financing
”).The participants in the GreenLight Series C Preferred Stock Financing included persons affiliated with members of GreenLight’s board of directors and persons that held more than 5% of GreenLight’s outstanding capital stock at the time of the closing of the GreenLight Series C Preferred Stock Financing. The following table summarizes purchases of shares of GreenLight Series C Preferred Stock from GreenLight by such related persons:
Name | Number of Shares | Total Purchase Price | ||||||
S2G Ventures Fund I, L.P. (1) | 3,135,582 | $ | 4,985,575 | |||||
S2G Ventures Fund II, L.P. (1) | 3,135,583 | $ | 4,985,576 | |||||
Baird Venture Partners V Limited Partnership (2) | 3,762,699 | $ | 5,982,691 | |||||
Fall Line Endurance Fund, LP (3) | 3,135,583 | $ | 4,985,576 | |||||
Kodiak Venture Partners III, L.P. (4) | 2,753,920 | $ | 4,378,733 | |||||
Kodiak III Entrepreneurs Fund, L.P. (4) | 68,104 | $ | 108,285 | |||||
Series Greenlight, a Separate Series of BlueIO Growth LLC (4) | 1,301,266 | $ | 2,074,999 | |||||
Furneaux Capital Holdco, LLC (dba BlueIO) (4) | 188,134 | $ | 299,998 | |||||
MLS Capital Fund II, L.P. (5) | 1,881,350 | $ | 2,991,357 | |||||
| (1) | Matthew Walker was a member of the GreenLight board of directors at the time of the GreenLight Series D Preferred Stock Financing. Mr. Walker joined the board of directors at the time of the closing of the GreenLight Series C Preferred Stock Financing and continuously served on the GreenLight board of directors since that time until the Closing of the Business Combination. S2G Ventures acquired more than 5% of GreenLight’s outstanding capital stock in the GreenLight Series C Preferred Stock Financing. Mr. Walker became a New GreenLight director upon the Closing of the Business Combination. |
| (2) | Michael Liang was a member of the GreenLight board of directors. Mr. Liang joined the board of directors at the time of the closing of the GreenLight Series C Preferred Stock Financing and continuously served on |
149
| the GreenLight board of directors since that time until the Closing of the Business Combination. During this period, he has been an affiliate of Baird. Baird acquired more than 5% of GreenLight’s outstanding capital stock in the GreenLight Series C Preferred Stock Financing. |
| (3) | Eric O’Brien was a member of the GreenLight board of directors at the time of the GreenLight Series C Preferred Stock Financing and continuously served on the GreenLight board of directors since that time until the Closing of the Business Combination. During this period, he has been an affiliate of Fall Line. Fall Line held more than 5% of GreenLight’s outstanding capital stock at the time of the GreenLight Series C Preferred Stock Financing and continuously held more than 5% of GreenLight’s outstanding capital stock at all times since the GreenLight Series C Preferred Stock Financing. Mr. O’Brien became a New GreenLight director upon the Closing of the Business Combination. |
| (4) | David Furneaux was a member of the GreenLight board of directors at the time of the GreenLight Series C Preferred Stock Financing. Mr. Furneaux joined the board of directors in June 2013 and served on the GreenLight board of directors until the initial closing of the GreenLight Series D Preferred Stock Financing. During this period, he was an affiliate of (i) Kodiak Venture Partners III, L.P. and Kodiak III Entrepreneurs |
Fund, L.P. (collectively, “
Kodiak
”), (ii) Series Greenlight 2 and Series Greenlight, a Separate Series of BlueIO Growth LLC (collectively, “Series Greenlight
”) and (iii) Furneaux Capital Holdco, LLC (dba BlueIO) (“Furneaux Capital
” and, together with Series Greenlight, “BlueIO
”). Kodiak acquired more than 5% of GreenLight’s outstanding capital stock in the GreenLight Series C Preferred Stock Financing. BlueIO held less than 5% of GreenLight’s outstanding capital stock at the time of GreenLight Series C Preferred Stock Financing.| (5) | Ganesh Kishore was a member of the GreenLight board of directors at the time of the GreenLight Series C Preferred Stock Financing and continuously served on the GreenLight board of directors since that time until the Closing of the Business Combination. During this period, he has been an affiliate of MLS. MLS held more than 5% of GreenLight’s outstanding capital stock at the time of the GreenLight Series C Preferred Stock Financing and has continuously held more than 5% of GreenLight’s outstanding capital stock at all times since the GreenLight Series C Preferred Stock Financing. Mr. Kishore became a New GreenLight director upon the Closing of the Business Combination. |
GreenLight Investors’ Rights Agreement
GreenLight was a party to a Fifth Amended and Restated Investors’ Rights Agreement, dated as of June 15, 2020 (the “
GreenLight IRA
”), which granted certain customary registration rights and information rights, among other things, to certain holders of GreenLight’s capital stock, including Morningside, Baird, Fall Line, MLS, S2G Ventures, Kodiak, and Khosla Ventures V, LP (“Khosla V
”), Khosla Ventures Seed B, LP (“Khosla Seed
” and, together with Khosla V, “Khosla
”) and entities affiliated with such persons. These stockholders became parties to the GreenLight IRA (or an earlier version thereof) in connection with their respective investments in GreenLight, including in connection with the GreenLight Series D Preferred Stock Financing and the GreenLight Series C Preferred Stock Financing, as applicable. The GreenLight IRA terminated upon the closing of the Business Combination.GreenLight Voting Agreement
GreenLight was a party to the Fifth Amended and Restated Voting Agreement, dated as of June 15, 2020 (the “
GreenLight Voting Agreement
”), pursuant to which certain holders of GreenLight’s capital stock, including Morningside, Baird, Fall Line, MLS, S2G Ventures, Kodiak, and Khosla and entities affiliated with such persons, agreed to vote their shares of GreenLight’s capital stock in favor of certain matters, including with respect to the election of directors. These stockholders became parties to the GreenLight Voting Agreement (or an earlier version thereof) in connection with their respective investments in GreenLight, including in connection with the GreenLight Series D Preferred Stock Financing and the GreenLight Series C Preferred Stock Financing, as applicable. The GreenLight Voting Agreement terminated upon the closing of the Business Combination.150
Management Rights Letter
GreenLight was a party to a Management Rights Letter with Morningside, dated as of June 15, 2020 (the “
Management Rights Letter
”), pursuant to which Morningside was granted certain customary information rights in connection with its investment in GreenLight. The Management Rights Letter terminated upon the closing of the Business Combination.Business Combination Arrangements
Certain of GreenLight’s stockholders, directors and executive officers entered into agreements with ENVI in connection with Business Combination. The agreements described in this section, or forms of such agreements, are filed as exhibits to this annual report, and the following descriptions are qualified by reference thereto. These agreements include:
| • | the Subscription Agreements, which were executed and delivered by the following holders of more than 5% of GreenLight’s outstanding capital stock or an affiliate thereof: S2G Builders Food & Agriculture Fund III, LP (an affiliate of Builders Vision, LLC), Morningside, Fall Line, and MLS; |
| • | the Transaction Support Agreements, which were executed and delivered by the following holders of more than 5% of GreenLight’s outstanding capital stock and directors and executive officers of GreenLight: Fall Line, Khosla, Kodiak, MLS, Morningside, S2G Ventures, and Drs. Andrey Zarur, Charles Cooney and Martha Schlicher; and |
| • | the Investor Rights Agreement, which was executed and delivered by the following holders of more than 5% of GreenLight’s outstanding capital stock and directors and executive officers of GreenLight: Fall Line, Khosla, Kodiak, MLS, Morningside, S2G Ventures and Dr. Andrey Zarur. |
These agreements are described in more detail below and in Exhibit 4.3, “
Description of Securities — Registration Rights — Investor Rights Agreement.”
Subscription Agreements for the PIPE Financing
Concurrently with the execution of the Business Combination Agreement and in November 2021, ENVI entered into the Subscription Agreements with each of the PIPE Investors, pursuant to which the PIPE Investors subscribed for and purchased, and ENVI issued and sold to the PIPE Investors, on the Closing Date immediately prior to the Closing, an aggregate of 12,425,000 shares of ENVI Class A Common Stock at a price of $10.00 per share, for aggregate gross proceeds of $124,525,000. The shares of ENVI Class A Common Stock issued pursuant to the Subscription Agreements were not registered under the Securities Act and were issued in reliance upon the exemption provided in Section 4(a)(2) of the Securities Act. The PIPE Financing was contingent upon, among other things, the substantially concurrent closing of the Business Combination. In connection with the Business Combination, all of the issued and outstanding shares of ENVI Class A Common Stock, including the shares of ENVI Class A Common Stock issued in the PIPE Financing, and all of the issued and outstanding ENVI Class B Common Stock became shares of New GreenLight Common Stock.
Subscription Agreements were executed by the following holders of GreenLight’s outstanding capital stock or an affiliate thereof, as follows:
Name | Number of Shares | Subscription Amount | ||||||
S2G Builders Food & Agriculture Fund III, LP (1) | 1,500,000 | $ | 15,000,000 | |||||
Morningside Venture Investments Limited | 1,000,000 | $ | 10,000,000 | |||||
Fall Line Endurance Fund, LP | 700,000 | $ | 7,000,000 | |||||
MLS Capital Fund II, L.P. | 75,000 | $ | 750,000 | |||||
| (1) | S2G Builders Food & Agriculture Fund III, LP is an affiliate of Builders Vision, LLC. |
151
In December 2021, the Prepaying PIPE Investors advanced an aggregate of $35,250,000 of the proceeds of the PIPE Financing to us through the purchase of convertible securities (the “”) that ENVI would accept a tender by a Prepaying PIPE Investor of its Instrument as payment of the purchase price under the Prepaying PIPE Investor’s Subscription Agreement in an amount equal to the outstanding principal and interest accrued on the Instrument as of the date of the closing under the Subscription Agreement. If the aggregate amount of such principal and interest exceeded the applicable purchase price under the Subscription Agreement, ENVI would pay the excess to the Prepaying PIPE Investor in cash. If the aggregate amount of such principal and interest was less than the applicable purchase price under the Subscription Agreement, the Prepaying PIPE Investor was obligated to pay the difference in cash in order to satisfy its obligations under the Subscription Agreement. GreenLight and ENVI also agreed that the aggregate amount of principal and accrued interest on the convertible instruments would be included for purposes of calculating the Aggregate Closing PIPE Proceeds (as defined in the Business Combination Agreement). GreenLight, ENVI and each Prepaying PIPE Investor agreed to treat the Instruments as equity interests and not as indebtedness for U.S. federal income tax purposes. At the Closing, ENVI accepted the surrender of Instruments as payment of the purchase price for an aggregate of 3,525,000 shares of ENVI Class A Common Stock in the PIPE Financing (including 2,775,000 shares being purchased by GreenLight stockholders or their affiliates) at a purchase price of $10.00 per share, and paid the holders of such Instruments aggregate interest of approximately $10,300 in cash (of which approximately $8,000 was payable to GreenLight stockholders and their affiliates). For more information about the advancement of the proceeds from the PIPE Financing, see the GreenLight MD&A under the subheading “”
Instruments
”) pursuant to the terms of a Convertible Instrument Investment Agreement (the “Investment Agreement
”) entered into between GreenLight and the Prepaying PIPE Investors. Of this amount, S2G Builders Food & Agriculture Fund III, LP, Fall Line Endurance Fund, LP, Morningside Venture Investments Limited and MLS Capital Fund II, L.P. advanced $15 million, $7 million, $5 million and $750,000, respectively. The Instruments provided that they would mature 12 months after the date of issuance (or, if earlier, upon an event of default specified in the Instruments) and bore interest at the minimum applicable federal rate per annum, which interest was payable at maturity. In connection with the issuance of the Instruments, GreenLight, ENVI and each Prepaying PIPE Investor agreed in a letter agreement (each such agreement, a “Letter Agreement
— Liquidity and Capital Resources — Advancement of a Portion of the Purchase Price of the PIPE Financing.
The Subscription Agreements grant the PIPE Investors certain resale registration rights in connection with the PIPE Financing. ENVI agreed to file a registration statement with the SEC within 30 days after the Closing to register the resale of the shares acquired in the PIPE Financing. ENVI agreed to use its commercially reasonable efforts to have the registration statement declared effective as soon as practicable after filing but no later than the earlier of (i) the 60th day (or 90th day if the SEC notifies ENVI that it will “review” the registration statement) following the Closing and (ii) the 10th business day after the date ENVI is notified by the SEC that the registration statement will not be “reviewed” or will not be subject to further review.
Transaction Support Agreements
Concurrently with the execution of the Business Combination Agreement, ENVI and certain stockholders of GreenLight (collectively, the “
Supporting Company Shareholders
”) entered into Transaction Support Agreements, pursuant to which each such holder agreed, subject to the terms and conditions of the Transaction Support Agreement, to (i) vote in favor of and consent to the Business Combination Agreement and the transactions contemplated thereby (including the Merger), (ii) waive his, her or its appraisal rights with respect to the Merger, (iii) effective immediately prior to, and contingent upon, the Effective Time, to terminate (a) the Fifth Amended and Restated Investors’ Rights Agreement, dated as of June 15, 2020, by and among GreenLight and the investor parties thereto, as amended, (b) the Fifth Amended and Restated Right of First Refusal andCo-Sale
Agreement, dated as of June 15, 2020, by and among GreenLight, the investor and key holder parties thereto, as amended, (c) the Fifth Amended and Restated Voting Agreement, dated as of June 15, 2020, by and among GreenLight, the investor and key holder parties thereto, as amended, and (d) any management rights letter, investor side letter or other investor agreement providing for board observer rights, information rights, inspection rights or other rights of investors, in each case between or among GreenLight and the Supporting152
Company Shareholder (and/or other persons), and all rights and obligations contained therein and (iv) be bound by certain other covenants and agreements related to the Business Combination, including an agreement to be bound by the exclusive dealing provisions of the Business Combination Agreement and restrictions on transfers with respect to his, her or its shares of capital stock of GreenLight prior to the closing of the Business Combination.
Investor Rights Agreement
For a description of the terms of the Investor Rights Agreement, see Exhibit 4.3, “.”
Description of Securities — Registration Rights — Rights Agreement
Indemnification under our Charter and Bylaws; Indemnification Agreements
The Charter and Bylaws provide that New GreenLight will indemnify its directors and officers to the maximum extent permitted by the DGCL, subject to certain exceptions. In addition, the Charter provides that our directors will not be liable for monetary damages for breach of fiduciary duty to the maximum extent permitted by the DGCL.
New GreenLight entered into indemnification agreements with each of its directors and executive officers. The indemnification agreements provide the indemnitees with contractual rights to indemnification, and expense advancement and reimbursement, to the maximum extent permitted under the DGCL, subject to certain exceptions contained in those agreements. For additional information, see Exhibit 4.3, “.”
Description of New GreenLight Securities—Limitations on Liability and Indemnification of Officers and Directors
Policies and Procedures for Related Party Transactions
The board of directors of New GreenLight adopted a written related person transaction policy that sets forth the following policies and procedures for the review and approval or ratification of related person transactions. A “related person transaction” is a transaction, arrangement or relationship in which New GreenLight or any of its subsidiaries was, is or will be a participant, the amount of which involved exceeds $120,000, and in which any related person had, has or will have a direct or indirect material interest. A “related person” means:
| • | any person who is, or at any time during the applicable period was, one of New GreenLight’s directors or executive officers; |
| • | any person who is known by New GreenLight to be the beneficial owner of more than 5% of its voting stock; |
| • | any immediate family member of any of the foregoing persons, which means any child, stepchild, parent, stepparent, spouse, sibling, mother-in-law, father-in-law, son-in-law, daughter-in-law, brother-in-law or sister-in-law of |
| • | any firm, corporation or other entity in which any of the foregoing persons is a partner or principal, or in a similar position, or in which such person has a 10% or greater beneficial ownership interest. |
New GreenLight has policies and procedures designed to minimize potential conflicts of interest arising from any dealings it may have with its affiliates and to provide appropriate procedures for the disclosure of any real or potential conflicts of interest that may exist from time to time. Specifically, pursuant to its audit committee charter, the audit committee has the responsibility to review related party transactions.
153
ITEM 14. | Principal Accountant Fees and Services |
ENVI
The following table provides a summary of the fees for professional services rendered by WithumSmith+Brown, PC, ENVI’s independent registered public accounting firm, for the year ended December 31, 2021 and the period from July 2, 2020 (inception) through December 31, 2020 and by Deloitte & Touche LLP, GreenLight’s independent registered public accounting firm, for the years ended December 31, 2021 and 2020:
Year Ended December 31, 2021 | Period from July 2, 2020 (Inception) to December 31, 2020 | |||||||
ENVI — WithumSmith+Brown, PC | ||||||||
Audit Fees (1) | $ | 63,860 | $ | 83,945 | ||||
Audit-Related Fees (2) | — | — | ||||||
Tax Fees (3) | — | $ | 7,725 | |||||
All Other Fees (4) | $ | 42,230 | — | |||||
Total | $ | 106,090 | $ | 91,670 | ||||
Year Ended December 31, 2021 | Year Ended December 31, 2020 | |||||||
GreenLight — Delotte & Touche LLP | ||||||||
Audit Fees (1) | $ | 2,074,967 | $ | 188,106 | ||||
Audit-Related Fees (2) | 1,895 | 1,895 | ||||||
Tax Fees (3) | — | — | ||||||
All Other Fees (4) | — | — | ||||||
Total | 2,076,862 | 190,001 | ||||||
| (1) | Audit fees consist of fees billed for professional services rendered for the audit of year-end consolidated financial statements, reviews of quarterly interim financial statements and services that are normally provided by the independent registered public accounting firm in connection with statutory and regulatory filings, as well as fees incurred in connection with the preparation and filing of registration statements with the SEC. The above amounts include interim procedures and audit fees, as well as attendance at audit committee meetings. |
| (2) | Audit-related fees consist of fees billed for assurance and related services that are reasonably related to performance of the audit or review of year-end consolidated financial statements and are not reported under “Audit Fees.” These services include attest services that are not required by statute or regulation and consultation concerning financial accounting and reporting standards. |
| (3) | Tax fees consist of fees billed for professional services relating to tax compliance, tax planning and tax advice. |
| (4) | All other fees consist of fees billed for all other services, including permitted due diligence services related to potential business combinations. |
Policy on Board
Pre-Approval
of Audit and PermittedNon-Audit
Services of the Registered Independent Public Accounting FirmENVI’s audit committee was formed upon the consummation of its initial public offering. As a result, its audit committee did not
pre-approve
all of the foregoing services, although any services rendered prior to the formation of the audit committee were approved by ENVI’s board of directors.Since the formation of ENVI’s audit committee, it has been the policy of the audit committee to
pre-approve
all auditing services and permittednon-audit
services to be performed for us by the independent registered public154
accounting firm, including the fees and terms thereof, subject to permitted de minimis exceptions for
non-audit
services which are approved by the audit committee prior to the completion of the audit. Neither WithumSmith+Brown, PC nor Deloitte & Touche LLP provided any services in reliance on such de minimis exception for any of the foregoing periods.155
PART IV
ITEM 15. Exhibits and Financial Statement Schedules
(a) | Exhibits |
156
157
158
| + | Indicates management contract or compensatory plan. |
| † | Certain identified information has been excluded from this exhibit because either (a) the information is both not material and the type of information that the Registrant treats as private or confidential or (b) disclosure of such information would constitute a clearly unwarranted invasion of personal privacy. |
| †† | Schedules and exhibits to this Exhibit omitted pursuant to Regulation S-K Item 601(a)(5). The Registrant agrees to furnish supplementally a copy of any omitted schedule of exhibit to the SEC upon request. |
(b) | Financial Statement Schedules. |
All financial statement schedules have been omitted because the information required to be set forth therein is not applicable or is shown in the financial statements or notes thereto.
ITEM 16. Form
10-K
SummaryNone.
159
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this annual report to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Medford, Massachusetts on the 31
st
day of March, 2022.GREENLIGHT BIOSCIENCES HOLDINGS, PBC | ||
| By: | /s/ Andrey J. Zarur | |
| Name: | Dr. Andrey J. Zarur | |
| Title: | Chief Executive Officer, President and Director | |
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints each of Andrey J. Zarur, Susan Keefe and David Kennedy as his or her true and lawfuland agent, with full power of substitution and resubstitution, for him or her and in his or her name, place and stead, in any and all capacities, to sign any and all amendments to this Annual Report on Formand agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that saidand agents, or any of them, or their or his or her substitutes or substitute, may lawfully do or cause to be done by virtue hereof.
attorney-in-fact
10-K,
and to file the same, with all exhibits thereto, and other documents in connection therewith, with the United States Securities and Exchange Commission, granting unto saidattorneys-in-fact
attorneys-in-fact
Pursuant to the requirements of the Securities Act of 1934, as amended, this annual report been signed below by the following person in the capacities and on the dates indicated.
Signature | Title | Date | ||
/s/ Dr. Andrey J. Zarur Dr. Andrey J. Zarur | President, Chief Executive Officer and Director | March 31, 2022 | ||
| (Principal Executive Officer) | ||||
/s/ Susan Keefe Susan Keefe | Chief Financial Officer and Interim Chief Accounting Officer | March 31, 2022 | ||
| (Principal Financial and Accounting Officer) | ||||
/s/ Dr. Charles Cooney | ||||
Dr. Charles Cooney | Director | March 31, 2022 | ||
/s/ Dr. Ganesh Kishore | ||||
Dr. Ganesh Kishore | Director | March 31, 2022 | ||
/s/ Eric O’Brien | ||||
Eric O’Brien | Director | March 31, 2022 | ||
160
Signature | Title | Date | ||
/s/ Jennifer E. Pardi | ||||
Jennifer E. Pardi | Director | March 31, 2022 | ||
/s/ Dr. Martha Schlicher | ||||
| Dr. Martha Schlicher | Director | March 31, 2022 | ||
/s/ Matthew Walker | ||||
Matthew Walker | Director | March 31, 2022 | ||
161
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
FORMERLY KNOWN AS ENVIRONMENTAL IMPACT ACQUISITION CORP.
INDEX TO FINANCIAL STATEMENTS
| F-2 | ||
Financial Statements: | ||
| F-3 | ||
| F-4 | ||
| F-5 | ||
| F-6 | ||
| F-7 |
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and the Board of Directors of
GreenLight Biosciences Holdings, PBC:
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of GreenLight Biosciences Holdings, PBC, formerly known as Environmental Impact Acquisition Corp. (the “Company”) as of December 31, 2021 and 2020 and the related consolidated statements of operations, changes in stockholders’ (deficit) equity and cash flows for the year ended December 31, 2021 and for the period from July 2, 2020 (inception) through December 31, 2020 and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2021 and 2020, and the results of its operations and its cash flows for the year ended December 31, 2021 and for the period from July 2, 2020 (inception) through December 31, 2020, in conformity with accounting principles generally accepted in the United States of America.
Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, if the Company is unable to raise additional funds to alleviate liquidity needs, then the Company will cease all operations except for the purpose of liquidating. The liquidity condition raises substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provides a reasonable basis for our opinion.
| /s/ WithumSmith+Brown, PC |
| We have served as the Company’s auditor since 2020. |
| New York, New York |
| March 31, 2022 |
| PCAOB ID Number 100 |
F-2
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
CONSOLIDATED BALANCE SHEETS
December 31, 2021 | December 31, 2020 | |||||||
ASSETS | ||||||||
Current assets | ||||||||
Cash | $ | 67,496 | $ | 156,848 | ||||
Prepaid expenses | 462,550 | — | ||||||
Total Current Assets | 530,046 | 156,848 | ||||||
Deferred offering costs | — | 168,152 | ||||||
| Marketable securities held in Trust Account | 207,012,391 | — | ||||||
TOTAL ASSETS | $ | 207,542,437 | $ | 325,000 | ||||
| LIABILITIES, CLASS A COMMON STOCK SUBJECT TO POSSIBLE REDEMPTION, AND STOCKHOLDERS’ (DEFICIT) EQUITY | ||||||||
Current liabilities | ||||||||
Accounts payable and accrued expenses | $ | 11,702,085 | $ | 2,528 | ||||
Accrued offering costs | 118,569 | — | ||||||
Promissory note – related party | 500,000 | 300,000 | ||||||
Total Current Liabilities | 12,320,654 | 302,528 | ||||||
Warrant liabilities | 15,885,000 | — | ||||||
Total Liabilities | 28,205,654 | 302,528 | ||||||
Commitments and Contingencies | 0 | 0 | ||||||
Class A common stock subject to possible redemption 20,700,000 and 0 shares at redemption value of $10.00 as of December 31, 2021 and 2020, respectively | 207,000,000 | — | ||||||
Stockholders’ (Deficit) Equity | ||||||||
Preferred stock, $0.0001 par value; 1,000,000 shares authorized; 0ne issued or outstanding | 0— | 0— | ||||||
Class A common stock, $0.0001 par value; 100,000,000 shares authorized | 0— | 0— | ||||||
Class B common stock, $0.0001 par value; 20,000,000 shares authorized; 5,175,000 shares issued and outstanding as of December 31, 2021 and 2020 | 518 | 518 | ||||||
Additional paid-in capital | — | 24,482 | ||||||
Accumulated deficit | (27,663,735 | ) | (2,528 | ) | ||||
Total Stockholders’ (Deficit) Equity | (27,663,217 | ) | 22,472 | |||||
| TOTAL LIABILITIES, CLASS A COMMON STOCK SUBJECT TO POSSIBLE REDEMPTION, AND STOCKHOLDERS’ (DEFICIT) EQUITY | $ | 207,542,437 | $ | 325,000 | ||||
The accompanying notes are an integral part of the consolidated financial statements.
F-3
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
CONSOLIDATED STATEMENTS OF OPERATIONS
Year Ended December 31, 2021 | For the Period from July 2, 2020 (Inception) Through December 31, 2020 | |||||||
General and administrative expenses | $ | 13,095,341 | $ | 2,528 | ||||
Loss from operations | (13,095,341 | ) | (2,528 | ) | ||||
Other expense: | ||||||||
Interest earned on marketable securities held in Trust Account | 12,391 | — | ||||||
Loss in initial issuance of Private Placement Warrants | (1,272,500 | ) | — | |||||
Change in fair value of warrant liabilities | (704,000 | ) | — | |||||
Other expense, net | (1,964,109 | ) | — | |||||
Net loss | $ | (15,059,450 | ) | $ | (2,528 | ) | ||
Weighted average shares outstanding, Class A common stock | $ | 19,679,178 | — | |||||
Basic and diluted net loss per share, Class A common stock | $ | (0.61 | ) | $ | — | |||
Weighted average shares outstanding, Class B common stock | 5,141,712 | 4,500,000 | ||||||
Basic and diluted net loss per share, Class B common stock | $ | (0.61 | ) | $ | (0.00 | ) | ||
The accompanying notes are an integral part of the consolidated financial statements.
F-4
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ (DEFICIT) EQUITY
Class A Common Stock | Class B Common Stock | Additional Paid-in | Accumulated | Total Stockholders’ Equity | ||||||||||||||||||||||||
Shares | Amount | Shares | Amount | Capital | Deficit | (Deficit) | ||||||||||||||||||||||
Balance — July 2, 2020 (inception) | 0— | $ | 0— | 0— | $ | 0— | $ | 0— | $ | 0— | $ | 0— | ||||||||||||||||
Issuance of Class B common stock to Sponsor | — | — | 5,175,000 | 518 | 24,482 | — | 25,000 | |||||||||||||||||||||
Net loss | — | — | — | — | — | (2,528 | ) | (2,528 | ) | |||||||||||||||||||
Balance — December 31, 2020 | 0— | $ | 0— | 5,175,000 | $ | 518 | $ | 24,482 | $ | (2,528 | ) | $ | 22,472 | |||||||||||||||
Accretion of Class A common stock subject to possible redemption | — | — | — | — | (24,482 | ) | (12,601,757 | ) | (12,626,239 | ) | ||||||||||||||||||
Net l o ss | — | — | — | — | — | (15,059,450 | ) | (15,059,450 | ) | |||||||||||||||||||
Balance – December 31, 2021 | 0— | $ | 0— | 5,175,000 | $ | 518 | $ | 0 | $ | (27,663,735 | ) | $ | (27,663,217 | ) | ||||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
F-5
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
CONSOLIDATED STATEMENTS OF CASH FLOWS
Year Ended December 31, 2021 | For the Period from July 2, 2020 (Inception) through December 31, 2020 | |||||||
Cash Flows from Operating Activities: | ||||||||
Net loss | $ | (15,059,450 | ) | $ | (2,528 | ) | ||
Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
Loss on issuance of Private Placement Warrants | 1,272,500 | — | ||||||
Change in fair value of warrant liabilities | 704,000 | — | ||||||
Transaction costs incurred in connection with warrants | 50,179 | — | ||||||
Interest earned on marketable securities held in Trust Account | (12,391 | ) | — | |||||
Changes in operating assets and liabilities: | ||||||||
Prepaid expenses | (462,550 | ) | — | |||||
Accounts payable and accrued expenses | 11,699,557 | 2,528 | ||||||
Net cash used in operating activities | (1,808,155 | ) | 0 | |||||
Cash Flows from Investing Activities: | ||||||||
Investment of cash into Trust Account | (207,000,000 | ) | — | |||||
Net cash used in investing activities | (207,000,000 | ) | 0 | |||||
Cash Flows from Financing Activities: | ||||||||
Proceeds from issuance of Class B common stock to Sponsor | — | 25,000 | ||||||
Proceeds from sale of Units, net of underwriting discounts paid | 206,750,000 | — | ||||||
Proceeds from sale of Private Placement Warrants | 2,000,000 | — | ||||||
Proceeds from sale of Unit Purchase Option | 6,000 | — | ||||||
Proceeds from promissory note - related party | 500,000 | — | ||||||
Repayment of promissory note - related party | (300,000 | ) | 180,632 | |||||
Payment of offering costs | (237,197 | ) | (48,784 | ) | ||||
Net cash provided by financing activities | 208,718,803 | 156,848 | ||||||
Net Change in Cash | (89,352 | ) | 156,848 | |||||
Cash – Beginning | 156,848 | — | ||||||
Cash – Ending | $ | 67,496 | $ | 156,848 | ||||
Non-cash investing and financing activities: | ||||||||
Offering costs included in accrued offering costs | $ | 118,569 | $ | — | ||||
Offering costs paid through promissory note | $ | — | $ | 119,368 | ||||
The accompanying notes are an integral part of the consolidated financial statements.
F-6
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
NOTE 1 — DESCRIPTION OF ORGANIZATION AND BUSINESS OPERATIONS
GreenLight Biosciences Holdings, PBC (formerly known as Environmental Impact Acquisition Corp.) (the “Company”) was incorporated in Delaware on July 2, 2020. The Company was formed for the purpose of effecting a merger, capital stock exchange, asset acquisition, stock purchase, reorganization or similar business combination with one or more businesses. The Company was not limited to a particular industry or sector for purposes of consummating a business combination. The Company is an early stage and emerging growth company and, as such, the Company is subject to all of the risks associated with early stage and emerging growth companies.
The Company has one wholly-owned subsidiary, Honey Bee Merger Sub, Inc., which was incorporated in the State of Delaware on August 6, 2021 (“Merger Sub”).
As of December 31, 2021, the Company had not commenced any operations. All activity for the period from July 2, 2020 (inception) through December 31, 2021, relates to the Company’s formation and the initial public offering (“Initial Public Offering”), which is described below, identifying a target company for a business combination and activities in connection with the acquisition of GreenLight Biosciences, Inc., a Delaware corporation (“GreenLight”) (see Note 6). The Company generated
non-operating
income in the form of interest income from the marketable securities held in the Trust Account.The registration statement for the Company’s Initial Public Offering was declared effective on January 13, 2021. On January 19, 2021 the Company consummated the Initial Public Offering of 20,700,000 units (the “Units” and, with respect to the Class A common stock included in the Units sold, the “Public Shares”), which includes the full exercise by the underwriter of its over-allotment option in the amount of 2,700,000 Units, at $10.00 per Unit, generating gross proceeds of $207,000,000 which is described in Note 3.
Simultaneously with the closing of the Initial Public Offering, the Company consummated the sale of 2,000,000 warrants (the “Private Placement Warrants”) at a price of $1.00 per Private Placement Warrant in a private placement to HB Strategies LLC (“HB Strategies”), the anchor investor and an affiliate of Hudson Bay Capital Management LP, generating gross proceeds of $2,000,000, which is described in Note 4.
Transaction costs amounted to $773,917, consisting of $250,000 in cash underwriting fees, inclusive of $150,000 paid for underwriters concession fees (see Note 6), and $523,917 of other offering costs.
Following the closing of the Initial Public Offering on January 19, 2021, an amount of $207,000,000 ($10.00
per Unit) from the net proceeds of the sale of the Units in the Initial Public Offering and the sale of the Private Placement Warrants was placed in a trust account (the “Trust Account”), located in the United States and was invested only in U.S. government securities, within the meaning set forth in Section 2(a)(16) of the Investment Company Act of 1940, as amended (the “Investment Company Act”) with a maturity of 185 days or less or in any open-ended investment company that holds itself out as a money market fund selected by the Company meeting certain conditions of Rule 2a-7 of the Investment Company Act, as determined by the Company, until the earlier of: (i) the completion of a business combination and (ii) the distribution of the funds held in the Trust Account, as described below.
The Company’s management has broad discretion with respect to the specific application of the net proceeds of the Initial Public Offering and the sale of Private Placement Warrants, although substantially all of the net proceeds have been applied toward consummating a business combination.
F-7
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
Consummation of Business Combination
On February 2, 2022 (the “Closing Date”), the Company consummated the previously announced business combination with GreenLight Biosciences, Inc., a Delaware corporation (“GreenLight”), pursuant to the terms of the business combination agreement, dated August 9, 2021 (the “Business Combination Agreement”), among the Company, GreenLight and Merger Sub. Pursuant to the terms of the Business Combination Agreement, Merger Sub merged with and into GreenLight, with GreenLight surviving the merger as a wholly owned subsidiary of ENVI (the “Merger” or “Business Combination”). In connection with the consummation of the Merger on the Closing Date, ENVI changed its name to GreenLight Biosciences Holdings, PBC (“New GreenLight”) and became a public benefit corporation.
In accordance with the terms and subject to the conditions of the Business Combination Agreement, at the effective time of the Merger (the “Effective Time”), each outstanding share of capital stock of GreenLight (other than treasury shares and shares with respect to which appraisal rights under the Delaware General Corporation Law (the “DGCL”) are properly exercised and not withdrawn) was exchanged for shares of common stock, par value $0.0001
per share, of New GreenLight (“New GreenLight Common Stock”), and outstanding GreenLight options and warrants to purchase shares of capital stock of GreenLight (whether vested or unvested) were converted into comparable options (the “Rollover Options”) and warrants (the “Assumed Warrants”) to purchase shares of New GreenLight Common Stock, in each case, based on an implied GreenLight fully diluted equity value
of $1.2 billion. In connection with the consummation of the Business Combination, all of the issued and outstanding shares of ENVI Class A common stock, par value $0.0001 per share (“ENVI Class A Common Stock”), and all of the issued and outstanding shares of ENVI Class B common stock, par value $0.0001per share (“ENVI Class B Common Stock”), became shares of New GreenLight Common Stock.
Consummation of PIPE Financing
In connection with the Business Combination, New GreenLight completed the sale and issuance of
12,425,000shares of New GreenLight Common Stock in a private placement at a purchase price of
$10.00per share pursuant to subscription agreements (the “Subscription Agreements”) that had been entered into between New GreenLight and certain institutional accredited investors (the “PIPE Investors”) either concurrently with the execution of the Business Combination Agreement or subsequently in November 2021 (the “PIPE Financing”).
Proceeds of the Business Combination and PIPE Financing
New GreenLight’s gross proceeds from the Business Combination and the PIPE Financing totaled approximately
$136.4 million, which included approximately
$12.1 million of funds held in New GreenLight’s trust account (after giving effect to redemptions) and approximately
$124.3 million of proceeds from the PIPE Financing (the “Aggregate Closing PIPE Proceeds”), inclusive of $35.25 million previously advanced to GreenLight,). The gross proceeds do not reflect estimated aggregate transaction expenses and other costs related to the Business Combination, the PIPE Financing and other transactions of approximately $25.0 million.Warrant Forfeiture
In connection with ENVI’s initial public offering, approximately
2,000,000 warrants (the “Private Placement Warrants”) were purchased by HB Strategies, LLC (“HB Strategies”) and an aggregate of 750,000warrants (the “Insider Warrants” and together with the Private Placement Warrants, the “Private Warrants”) were issued to ENVI’s sponsor, CG Investments Inc. VI (the “Sponsor”) and certain of ENVI’s directors (together with HB Strategies and the Sponsor, the “Initial Stockholders”). Concurrently with the execution of the Business Combination Agreement, the Initial Stockholders and GreenLight entered into an agreement (the “Sponsor Letter
F-8
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
Agreement”), pursuant to which HB Strategies and the Sponsor agreed that, if more than
25% of the shares of ENVI Class A Common Stock were redeemed pursuant to ENVI’s Amended and Restated Certificate of Incorporation in effect prior to the Business Combination (the “Existing Charter”), then they would forfeit 25% of the warrants issued to the Initial Stockholders. At the closing of the Business Combination (the “Closing”), pursuant to the terms of the Sponsor Letter Agreement, HB Strategies and the Sponsor forfeited an aggregate of 687,500 Private Placement Warrants and Insider Warrants.Liquidity and Going Concern
As of December 31, 2021, the Company had $67,496 in cash and a working capital deficit of $11,790,608.
The Company’s liquidity needs prior to the consummation of the Initial Public Offering were satisfied through the payment of $25,000 from the Sponsor to purchase the Founder Shares (as defined in Note 6), and loan proceeds from the Sponsor of $300,000 and $500,000 under separate Promissory Notes (as defined in Note 6). The Company repaid the $300,000 Note in full on January 19, 2021. The $500,000 Note was initiated on August 9, 2021 and total borrowings as of December 31, 2021 were $500,000. Subsequent to the consummation of the Initial Public Offering, the Company’s liquidity has been satisfied through the net proceeds from the consummation of the Initial Public Offering and the private placement held outside of the Trust Account.
On February 2, 2022, the Company consummated the Business Combination with GreenLight. The Company believes that the net proceeds from the Business Combination and the PIPE Financing, together with the combined entities existing cash and cash equivalents, will not be sufficient to fund its operations for twelve months from the date hereof.
Based on the net proceeds from the Business Combination and the PIPE Financing, together with existing cash and cash equivalents, the Company is evaluating a range of opportunities to extend cash runway, including management of program spending, platform licensing collaborations and potential financing activities.
The combined entity expects to incur significant expenses and operating losses for the foreseeable future as the Company advances product candidates through preclinical and clinical development and field trials, seek regulatory approval and pursue commercialization of any approved product candidates. It is expected that research and development and general and administrative costs will increase in connection with the Company’s planned research and development activities. Because of the numerous risks and uncertainties associated with research, development and commercialization of our product candidates, the Company is unable to estimate the exact amount of our working capital requirements. Future capital requirements will depend on many factors.
Until the combined Company can generate product revenues to support its cost structure, if any, the Company expects to finance cash needs through a combination of equity offerings, debt financings, collaborations and other similar arrangements. To the extent that additional capital is raised through the sale of equity or convertible securities, the ownership interest of stockholders will be or could be diluted, and the terms of these securities may include liquidation, dividend, redemption and other preferences that adversely affect the rights of the Company’s common stockholders. Debt financing and equity financing, if available, may involve agreements that include covenants limiting or restricting the ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If the Company raises funds through collaborations, or other similar arrangements with third parties, the Company may have to relinquish valuable rights to its technologies, future revenue streams, research programs or product candidates or grant licenses on terms that may not be favorable to the Company and/or may reduce the value of common stock. If the Company is unable to raise additional funds through equity or debt financings when needed, the Company may be required to delay, limit, reduce or terminate product development or future commercialization efforts or grant rights to
F-9
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
develop and market product candidates even if the Company would otherwise prefer to develop and market such product candidates directly.
As a result of the above, in connection with the Company’s assessment of going concern considerations in accordance with Financial Accounting Standard Board’s Accounting Standards Update (“ASU”)
2014-15,
“Disclosures of Uncertainties about an Entity’s Ability to Continue as a Going Concern,” management of the Company has determined that the liquidity condition raises substantial doubt about the Company’s ability to continue as a going concern through a year after the financial statements are issued. These financial statements do not include any adjustments relating to the recovery of the recorded assets or the classification of the liabilities that might be necessary should the Company be unable to continue as a going concern.Risks and Uncertainties
Management of the Company continues to evaluate the impact of the
COVID-19
global pandemic and has concluded that while it is reasonably possible that the virus could have a negative effect on the Company’s financial position, its results of operations, the specific impact is not readily determinable as of the date of these financial statements. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.NOTE 2 — SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation
The accompanying financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”) and pursuant to the rules and regulations of the U.S. Securities and Exchange Commission (the “SEC”).
Principles of Consolidation
The accompanying consolidated financial statements include the accounts of the Company and its wholly owned subsidiary. All significant intercompany balances and transactions have been eliminated in consolidation.
Emerging Growth Company
The Company is an “emerging growth company,” as defined in Section 2(a) of the Securities Act, as modified by the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”), and it may take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies including, but not limited to, not being required to comply with the independent registered public accounting firm attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, reduced disclosure obligations regarding executive compensation in its periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved.
Further, Section 102(b)(1) of the JOBS Act exempts emerging growth companies from being required to comply with new or revised financial accounting standards until private companies (that is, those that have not had a Securities Act registration statement declared effective or do not have a class of securities registered under the Exchange Act) are required to comply with the new or revised financial accounting standards. The JOBS Act provides that a company can elect to opt out of the extended transition period and comply with the requirements that apply to
non-emerging
growth companies but any such election to opt out is irrevocable. The Company has elected not to opt out of such extended transition period which means that when a standard is issued or revised and it has different application dates for public or private companies, the Company, as an emerging growthF-10
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
company, can adopt the new or revised standard at the time private companies adopt the new or revised standard. This may make comparison of the Company’s financial statements with another public company which is neither an emerging growth company nor an emerging growth company which has opted out of using the extended transition period difficult or impossible because of the potential differences in accounting standards used.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires the Company’s management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period.
Making estimates requires management to exercise significant judgment. It is at least reasonably possible that the estimate of the effect of a condition, situation or set of circumstances that existed at the date of the financial statements, which management considered in formulating its estimate, could change in the near term due to one or more future confirming events. One of the more significant accounting estimates included in these consolidated financial statements is the determination of the fair value of the warrant liabilities. Such estimates may be subject to change as more current information becomes available and accordingly the actual results could differ significantly from those estimates.
Class A Common Stock Subject to Possible Redemption
The Company accounts for its Class A common stock subject to possible redemption in accordance with the guidance in Accounting Standards Codification (“ASC”) Topic 480 “Distinguishing Liabilities from Equity.” Shares of Class A common stock subject to mandatory redemption are classified as a liability instrument and is measured at redemption value. Conditionally redeemable common stock (including common stock that features redemption rights that is either within the control of the holder or subject to redemption upon the occurrence of uncertain events not solely within the Company’s control) is classified as temporary equity. At all other times, common stock is classified as stockholders’ equity. The Company’s Class A common stock features certain redemption rights that are considered to be outside of the Company’s control and subject to occurrence of uncertain future events. Accordingly, at December 31, 2021 and 2020, Class A common stock subject to possible redemption is presented as temporary equity, outside of the stockholders’ (deficit) equity section of the Company’s consolidated balance sheets.
The Company recognizes changes in redemption value immediately as they occur and adjusts the carrying value of redeemable common stock to equal the redemption value at the end of each reporting period.
At December 31, 2021, the Class A common stock subject to possible redemption reflected in the consolidated balance sheet are reconciled in the following table:
Gross proceeds | $ | 207,000,000 | ||
Less: | ||||
Proceeds allocated to Public Warrants | (11,902,500 | ) | ||
Class A common stock issuance costs | (723,739 | ) | ||
Plus: | ||||
Accretion of carrying value to redemption value | 12,626,239 | |||
Class A common stock subject to possible redemption | $ | 207,000,000 | ||
F-11
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
Warrant Liabilities
The Company accounts for the Public Warrants (as defined in Note 3) and Private Placement Warrants (together, with the Public Warrants, the “Warrants”) in accordance with the guidance contained in ASC 815 under which the Warrants do not meet the criteria for equity treatment and must be recorded as liabilities. Accordingly, the Company classifies the Warrants as liabilities at their fair value and adjusts the Warrants to fair value at each reporting period. This liability is subject to
re-measurement
at each balance sheet date until exercised, and any change in fair value is recognized in the statements of operations. The Private Placement Warrants for periods where no observable traded price was available were valued using a Modified Black Scholes Option Pricing Model. The Public Warrants for periods where no observable traded price was available were valued using a Monte Carlo simulation. For periods subsequent to the detachment of the Public Warrants from the Units, the Public Warrant quoted market price was used as the fair value as of each relevant date.Income Taxes
The Company follows the asset and liability method of accounting for income taxes under ASC 740, “Income Taxes.” Deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to differences between the financial statements carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that included the enactment date. Valuation allowances are established, when necessary, to reduce deferred tax assets to the amount expected to be realized. As of December 31, 2021 and December 31, 2020, the Company had deferred tax assets with a full valuation allowance recorded against them.
ASC 740 prescribes a recognition threshold and a measurement attribute for the financial statement recognition and measurement of tax positions taken or expected to be taken in a tax return. For those benefits to be recognized, a tax position must be more likely than not to be sustained upon examination by taxing authorities. The Company recognizes accrued interest and penalties related to unrecognized tax benefits as income tax expense. There were no unrecognized tax benefits and no amounts accrued for interest and penalties as of December 31, 2021 and 2020. The Company is currently not aware of any issues under review that could result in significant payments, accruals or material deviation from its position. The Company is subject to income tax examinations by major taxing authorities since inception.
The Company’s currently taxable income primarily consists of interest income on the Trust Account. The Company’s general and administrative costs are generally considered
start-up
costs and are not currently deductible. During the year ended December 31, 2021, the Company recorded no income tax expense. The Company’s effective tax rate for the year ended December 31, 2021 was approximately 0%, which differs from the expected income tax rate mainly due to the change in the fair value of the warrant liabilities and thestart-up
costs (discussed above) which are not currently deductible.Net Loss Per Common Share
The Company complies with accounting and disclosure requirements of FASB ASC Topic 260, “Earnings Per Share”. Net income (loss) per common share is computed by dividing net income (loss) by the weighted average number of common shares outstanding for the period. The Company has two classes of stock, which are referred to as Class A common stock and Class B common stock. Income and losses are shared pro rata between the two classes of stock.. Accretion associated with the redeemable shares of Class A common stock is excluded from income (loss) per common share as the redemption value approximates fair value.
F-12
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
The calculation of diluted income (loss) per common share does not consider the effect of the warrants issued in connection with the (i) Initial Public Offering, and (ii) the private placement since the exercise of the warrants is contingent upon the occurrence of future events. The warrants are exercisable to purchase 13,100,000 shares of Class A common stock in the aggregate. As of December 31, 2021 and 2020, the Company did not have any dilutive securities or other contracts that could, potentially, be exercised or converted into common stock and then share in the earnings of the Company. As a result, diluted net income (loss) loss per common share is the same as basic net income (loss) per common share for the periods presented.
The following table reflects the calculation of basic and diluted net income (loss) per common share (in dollars, except per share amounts):
Year Ended December 31, 2021 | For the Period from July 2, 2020 (Inception) Through December 31, 2020 | |||||||||||||||
Class A | Class B | Class A | Class B | |||||||||||||
Basic and diluted net loss per common share | ||||||||||||||||
Numerator: | ||||||||||||||||
Allocation of net loss, as adjusted | $ | (11,939,846 | ) | $ | (3,119,604 | ) | $ | 0— | $ | (2,528 | ) | |||||
Denominator: | ||||||||||||||||
Basic and diluted weighted average shares outstanding | 19,679,178 | 5,141,712 | 0 | 4,500,000 | ||||||||||||
Basic and diluted net loss per common share | $ | (0.61 | ) | $ | (0.61 | ) | $ | 0— | $ | (0.00 | ) | |||||
Fair Value of Financial Instruments
The fair value of the Company’s assets and liabilities, which qualify as financial instruments under ASC Topic 820, “Fair Value Measurement,” approximates the carrying amounts represented in the Company’s balance sheet, primarily due to their short-term nature, except for the warrant liabilities (see Note 10).
Recent Accounting Standards
In August 2020, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”)
2020-06
— “Contracts in Entity’s Own Equity (Subtopic815-40)
(“ASU2020-06”)”,
to simplify accounting for certain financial instruments ASU2020-06
eliminates the current models that require separation of beneficial conversion and cash conversion features from convertible instruments and simplifies the derivative scope exception guidance pertaining to equity classification of contracts in an entity’s own equity. The new standard also introduces additional disclosures for convertible debt and freestanding instruments that are indexed to and settled in an entity’s own equity. ASU2020-06
amends the diluted earnings per share guidance, including the requirement to use theif-converted
method for all convertible instruments. ASU2020-06
is effective January 1, 2022 and should be applied on a full or modified retrospective basis, with early adoption permitted beginning on January 1, 2021. The Company is currently assessing the impact, if any, that ASU2020-06
would have on its financial position, results of operations or cash flows.Management does not believe that any recently issued, but not yet effective, accounting standards, if currently adopted, would have a material effect on the Company’s financial statements.
F-13
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
NOTE 3 — INITIAL PUBLIC OFFERING
Pursuant to the Initial Public Offering, the Company sold 20,700,000 Units, which includes a full exercise by the underwriters of their over-allotment option in the amount of 2,700,000 Units, at a price of $10.00 per Unit. Each Unit consists of one share of Class A common stock and
one-half
of one redeemable warrant (“Public Warrant”). Each whole Public Warrant entitles the holder to purchase one share of Class A common stock at a price of $11.50 per share, subject to adjustment (see Note 8).NOTE 4 — PRIVATE PLACEMENT
Simultaneously with the closing of the Initial Public Offering, HB Strategies and/or its affiliates purchased an aggregate of 2,000,000 Private Placement Warrants at a price of $1.00 per Private Placement Warrant ($2,000,000 in the aggregate) from the Company in a private placement. Each Private Placement Warrant will be exercisable to purchase one share of Class A common stock at a price of $11.50 per share, subject to adjustment (see Note 7). The proceeds from the sale of the Private Placement Warrants were added to the net proceeds from the Initial Public Offering held in the Trust Account. If the Company does not complete a Business Combination within the Combination Period, the proceeds from the sale of the Private Placement Warrants held in the Trust Account will be used to fund the redemption of the Public Shares (subject to the requirements of applicable law) and the Private Placement Warrants will expire worthless.
NOTE 5 — RELATED PARTY TRANSACTIONS
Founder Shares
In August and September of 2020, the Company issued an aggregate of 7,187,500 shares of Class B common stock (the “Founder Shares”) to the Sponsor and HB Strategies (together, the “Initial Stockholders”) for an aggregate price of $25,000. In December 2020, the Sponsor and HB Strategies returned to the Company, at no cost, 862,500 and 2,443,750 Founder Shares, respectively, and the Company issued an aggregate of 431,250 Founder Shares to its independent director nominees, resulting in an aggregate of 4,312,500 Founder Shares issued and outstanding. On January 13, 2021, the Company effected a stock dividend of 1.2 shares for each share of common stock outstanding, resulting in the Initial Stockholders holding an aggregate of 5,175,000 Founder Shares. The Founder Shares included an aggregate of up to 675,000 shares subject to forfeiture to the extent that the underwriters’ over-allotment is not exercised in full or in part, so that the number of Founder Shares will equal, on an
as-converted
basis, approximately 20% of the Company’s issued and outstanding ordinary shares after the Initial Public Offering. As a result of the underwriters’ election to fully exercise their over-allotment option, no Founder Shares are currently subject to forfeiture.The Initial Stockholders have agreed, subject to limited exceptions, not to transfer, assign or sell any of the Founder Shares until the earlier to occur of: (A) six months after the completion of a Business Combination and (B) subsequent to a Business Combination, (x) if the last sale price of the Class A common stock equals or exceeds $12.00 per share (as adjusted for stock splits, stock capitalizations, reorganizations, recapitalizations and the like) for any 20 trading days within any
30-trading
day period commencing at least 60 days after a Business Combination, or (y) the date on which the Company completes a liquidation, merger, capital stock exchange or other similar transaction that results in all of the Public Stockholders having the right to exchange their shares of common stock for cash, securities or other property.Promissory Note — Related Party
On September 4, 2020, HB Strategies issued an unsecured promissory note to the Company (the “Promissory Note”), pursuant to which the Company may borrow up to an aggregate principal amount of
F-14
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
$300,000. The Promissory Note is
non-interest
bearing and payable on the earlier of (i) March 31, 2021 or (ii) the consummation of the Initial Public Offering. As of December 31, 2020, there was $300,000 in borrowings outstanding under the Promissory Note, which was repaid at the closing of the Initial Public Offering on January 19, 2021. Borrowings under the Promissory Note are no longer available.On August 9, 2021, the Sponsor agreed to loan the Company an aggregate of up to $500,000 pursuant to a promissory note (the “Note”). The Note is
non-interest
bearing and payable upon consummation of the Company’s initial Business Combination. At December 31, 2021, there was $500,000 of borrowings under the Note.Related Party Loans
In order to finance transaction costs in connection with a Business Combination, the Sponsor, members of the Company’s management team or any of their respective affiliates or other third parties may, but are not obligated to, loan the Company funds as may be required (“Working Capital Loans”), which will be repaid only upon the consummation of a Business Combination. If the Company does not consummate a Business Combination, the Company may use a portion of any funds held outside the Trust Account to repay the Working Capital Loans; however, no proceeds from the Trust Account may be used for such repayment. If such funds are insufficient to repay the Working Capital Loans, the unpaid amounts would be forgiven. Up to $1,500,000 of the Working Capital Loans may be converted into units at a price of $10.00 per unit at the option of the holder. The units would be identical to the Placement Units. The warrants would be identical to the Private Placement Warrants. As of December 31, 2021 and 2020, there were no Working Capital Loans outstanding.
Sponsor and Director Compensation
At the closing of the Initial Public Offering, the Company issued 600,000 private placement-equivalent warrants to the Sponsor and 50,000 private placement-equivalent warrants to each of Gov. Patrick, Messrs. Brewster and Seavers, the Company’s independent director. Such warrants were issued for nominal amount and are identical to the Private Placement Warrants, including as to exercise price, exercisability and exercise period. The Company recorded the fair value of these warrants of approximately $0.9 million on the date of issuance which is included in loss on initial issuance of Private Placement Warrants in the statements of operations for the year ended December 31, 2021.
Underwriter
The underwriter is an affiliate of the Sponsor (see note 6).
NOTE 6 — COMMITMENTS AND CONTINGENCIES
Registration Rights
Pursuant to a registration rights agreement entered into on January 13, 2021, the holders of the Founder Shares, Private Placement Warrants and warrants that may be issued upon conversion of Working Capital Loans (and any Class A common stock issuable upon the exercise of the Private Placement Warrants and warrants that may be issued upon conversion of Working Capital Loans and upon conversion of the Founder Shares) will be entitled to registration rights pursuant to a registration rights agreement requiring the Company to register such securities for resale (in the case of the Founder Shares, only after conversion to our Class A common stock). The holders of the majority of these securities are entitled to make up to three demands, excluding short form demands, that the Company register such securities. In addition, the holders have certain “piggy-back”
F-15
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
registration rights with respect to registration statements filed subsequent to the completion of a Business Combination and rights to require the Company to register for resale such securities pursuant to Rule 415 under the Securities Act. However, the registration rights agreement provides that the Company will not permit any registration statement filed under the Securities Act to become effective until termination of the applicable
fivelock-up
period. The Company will bear the expenses incurred in connection with the filing of any such registration statements. Notwithstanding the foregoing, the Initial Stockholders may not exercise their demand and “piggyback” registration rights afterand
seven years, respectively, after the effective date of the Initial Public Offering and may not exercise its demand rights on more than one occasion.
In addition, pursuant to a registration agreement with Hudson Bay Capital Management LP (“Hudson Bay”) and its permitted transferees, the Company is required to register (i) resale of any securities purchased in the Initial Public Offering by filing a registration statement within 30 days after the closing of the Initial Public Offering and use its best effort to have such registration statement declared effective within 90 days after the closing of the Initial Public Offering; and (ii) resale of any Private Placement Warrants and shares of Class A common stock underlying the Private Placement Warrants by filing a registration statement within 30 days after the completion of a Business Combination and use its best effort to have such registration statement declared effective within 90 days after the completion of a Business Combination. In the event of any delay in filing and/or effectiveness of any aforesaid registration statement under the registration agreement with Hudson Bay and its permitted transferees, the unavailability of such restatement after effectiveness or a public information failure (each, a “Registration Default”), Hudson Bay and its permitted transferees are entitled to payments from the Company equal to 2% of the purchase price on the occurrence of each Registration Default and 2% per month (or a portion thereof pro rata) that such Registration Default continues to exist.
Underwriting Agreement
The Company also engaged a qualified independent underwriter to participate in the preparation of the registration statement and exercise the usual standards of “due diligence” in respect thereto. The Company paid the independent underwriter a fee of $100,000 upon the completion of the Initial Public Offering in consideration for its services and expenses as the qualified independent underwriter. Additionally, the Company agreed to pay the underwriter $150,000 in expenses to cover seller’s concessions to selling group member in connection with the Initial Public Offering. The independent underwriter will receive no other compensation.
Business Combination Marketing Agreement
The Company engaged Canaccord Genuity LLC (“Canaccord”) as advisors in connection with its Business Combination to assist the Company in arranging meetings with its stockholders to discuss the potential Business Combination and the target business’ attributes, introduce the Company to potential investors that may be interested in purchasing the Company’s securities, assist the Company in obtaining stockholder approval for the Business Combination and assist the Company with the preparation of its press releases and public filings in connection with the Business Combination. The Company will pay Canaccord for such services upon the consummation of a Business Combination a cash fee in an amount equal to 3.76 % of the gross proceeds of the Initial Public Offering if the underwriters’ over-allotment option is exercised in full. Pursuant to the terms of the business combination marketing agreement, no fee will be due if the Company does not complete a Business Combination. As of December 31, 2021, Cannacord is owed a total of $5,839,808 as a result of the closing of the
Business Combination
per the Business Combination Marketing Agreement.Business Combination and Related Agreements
ENVI held a special meeting of the ENVI stockholders on February 1, 2022 (the “Special Meeting”). At the Special Meeting, the stockholders considered and adopted, among other matters, the Business Combination
F-16
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
Agreement. On February 2, 2022, the parties to the Business Combination Agreement consummated the Business Combination.
Prior
to the Special Meeting, certain stockholders of ENVI exercised their right to redeem approximately 19.5 million shares of ENVI Class A Common Stock for cash at a price of approximately $10.00 per share, or aggregate payments of approximately $194.9 million. The redemption price was paid out of ENVI’s trust account.
On the Closing Date, the following transactions were completed:
| • | Merger Sub merged with and into GreenLight, with GreenLight surviving as a wholly owned subsidiary of New GreenLight; |
| • | ENVI filed an amended and restated certificate of incorporation, became a public benefit corporation under the Delaware General Corporation Law and changed its name to “GreenLight Biosciences Holdings, PBC”, and the Board adopted amended and restated bylaws; |
| • | All outstanding shares of capital stock of GreenLight (other than treasury shares and shares with respect to which appraisal rights under the Delaware General Corporation Law are properly exercised and not withdrawn) were exchanged for an aggregate of approximately 104.0 million shares of New GreenLight Common Stock; |
| • | all outstanding options to acquire shares of capital stock of GreenLight were assumed by New GreenLight and converted into options under the GreenLight Biosciences Holdings, PBC 2022 Equity and Incentive Plan (the “2022 Plan million shares of New GreenLight Common Stock (“ Rollover Options ”); |
| • | all outstanding warrants to acquire shares of capital stock of GreenLight were assumed by New GreenLight and converted into warrants to acquire an aggregate of 75,920shares of New GreenLight Common Stock (“Assumed Warrants |
| • | New GreenLight issued 12,425,000 shares ofNew GreenLight Common Stock in the PIPE Financing; and |
| • | the 5,175,000 outstanding shares of ENVI Class B Common Stock converted into 5,175,000 shares of New GreenLight Common Stock. |
Private Placement
In
connection with ENVI’s initial public offering, approximately 2,000,000 warrants (the “Private Placement Warrants”) were purchased by HB Strategies, LLC (“HB Strategies”) and an aggregate of 750,000 warrants (the “Insider Warrants” and together with the Private Placement Warrants, the “Private Warrants”) were issued to ENVI’s sponsor, CG Investments Inc. VI (the “Sponsor”) and certain of ENVI’s directors (together with HB Strategies and the Sponsor, the “Initial Stockholders”). Concurrently with the execution of the Business Combination Agreement, the Initial Stockholders and GreenLight entered into an agreement (the “Sponsor Letter Agreement”), pursuant to which HB Strategies and the Sponsor agreed that, if more than 25% of the shares of ENVI Class A Common Stock were redeemed pursuant to ENVI’s Amended and Restated Certificate of Incorporation in effect prior to the Business Combination (the “Existing Charter”), then they would forfeit 25% of the warrants issued to the Initial Stockholders. At the closing of the Business Combination (the “Closing”), pursuant to the terms of the Sponsor Letter Agreement, HB Strategies and the Sponsor forfeited an aggregate of 687,500 Private Placement Warrants and Insider Warrants.F-17
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
Registration Rights and Transfer Restrictions
Concurrently with the execution of the Business Combination Agreement, ENVI, the initial stockholders and certain stockholders of GreenLight entered into the Investor Rights Agreement, dated August 9, 2021 (the “.”
Investor Rights Agreement
”), pursuant to which, among other things, the initial stockholders and such stockholders of GreenLight agreed not to effect any sale or distribution of any equity securities of ENVI duringthe lock-up period
described therein and will be granted certain registration rights, in each case subject to, and conditioned upon and effective as of, the effective time of the Merger. For additional information, see “The
Business Combination Proposal—Related Agreements—Investor Rights Agreement
Transaction Support Agreement
Concurrently with the execution of the Business Combination Agreement, ENVI and certain stockholders of GreenLight entered into the Transaction Support Agreement, dated August 9, 2021 (the “
Transaction Support
Agreement
”), pursuant to which each such holder agreed, subject to the terms and conditions of the Transaction Support Agreement, to (i) vote in favor of and consent to the Business Combination Agreement and the transactions contemplated thereby (including the Merger), (ii) waive his, her or its appraisal rights with respect to the Merger, (iii) effective immediately prior to, and contingent upon, the Effective Time, to terminate (a) the Fifth Amended and Restated Investors’ Rights Agreement, dated as of June 15, 2020, by and among GreenLight and the investor parties thereto, as amended, (b) the Fifth Amended and Restated Right of First Refusaland Co-Sale Agreement,
dated as of June 15, 2020, by and among the Company, the investor and key holder parties thereto, as amended, (c) the Fifth Amended and Restated Voting Agreement, dated as of June 15, 2020, by and among the Company, the investor and key holder parties thereto, as amended, and (d) any management rights letter, investor side letter or other investor agreement providing for board observer rights, information rights, inspection rights or other rights of investors, in each case between or among the Company and such holder (and/or other persons), and all rights and obligations contained therein and (iv) be bound by certain other covenants and agreements related to the Business Combination, including an agreement to be bound by the exclusive dealing provisions of the Business Combination Agreement and restrictions on transfers with respect to his, her or its securities of GreenLight capital stock prior to the closing of the Business Combination. For additional information, see “The
Business Combination Proposal—Related
Agreements—Transaction Support Agreement
The approval of the Business Combination Agreement and the transactions contemplated thereby (including the Merger) by the GreenLight stockholders will require the approval of the holders of (a) a majority of the voting power of the outstanding GreenLight Shares, voting together on an as-converted basis, (b) a majority of the outstanding shares of GreenLight Common Stock, (c) a majority of the outstanding shares of GreenLight Preferred Stock, voting together on an as-converted basis, (d) a majority of the outstanding shares of GreenLight Series C Preferred Stock, voting as a separate class, and (e) a majority of the outstanding shares of GreenLight Series D Preferred Stock, voting as a separate class. The parties to the Transaction Support Agreement
hold in the aggregate (a)
92,514,094GreenLight Shares, or
64% of the total GreenLight Shares, (b)
1,846,446shares of GreenLight Common Stock, or
54% of the total outstanding shares of GreenLight Common Stock, (c)
90,667,648shares of GreenLight Preferred Stock on an
as-converted
basis, representing
64% of the total outstanding shares of GreenLight Preferred Stock on an
as-converted
basis, (d)
19,362,221shares of GreenLight Series C Preferred Stock, representing
55% of the total outstanding shares of GreenLight Series C Preferred Stock and (e)
37,021,189shares of GreenLight Series D Preferred Stock, representing
62% of the total outstanding shares of GreenLight Series D Preferred Stock. Accordingly, it is expected that, as of the relevant record date for the vote, the parties to the Transaction Support Agreement
will
hold in the aggregate a sufficient number of GreenLight Shares to provide the requisite approval from the GreenLight stockholders of the Business Combination Agreement and the transactions contemplated thereby (including the Merger).
F-18
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
NOTE 7 — STOCKHOLDERS’ EQUITY
Preferred Stock
Class
A Common Stock
Class
B Common Stock
Holders of Class A common stock and holders of Class B common stock will vote together as a single class on all matters submitted to a vote of the stockholders except as otherwise required by law.
The shares of Class B common stock will automatically convert into Class A common stock at the time of the Business Combination, on abasis, subject to adjustment. In the case that additional shares of Class A common stock, or equity-linked securities, are issued or deemed issued in connection with a Business Combination, the number of shares of Class A common stock issuable upon conversion of all Founder Shares will equal, in the aggregate, on anbasis.
one-for-one
as-converted
basis, 20% of the total number of shares of Class A common stock outstanding after such conversion (after giving effect to any redemptions of shares of Class A common stock by public stockholders), including the total number of shares of Class A common stock issued, or deemed issued or issuable upon conversion or exercise of any equity-linked securities or rights issued or deemed issued, by the Company in connection with or in relation to the consummation of a Business Combination, excluding (i) any shares of Class A common stock or equity-linked securities or rights exercisable for or convertible into shares of Class A common stock issued, or to be issued, to any seller in a Business Combination, (ii) any securities issued to the initial stockholders of the Company upon conversion of Working Capital Loans and (iii) any public shares redeemed by public stockholders in connection with a Business Combination, provided that such conversion of Founder Shares will never occur on a less thanone-for-one
NOTE 8 — WARRANTS
As of December 31, 2021, there were 10,350,000 Public Warrants outstanding. As of December 31, 2020, there were no Public Warrants outstanding. Public Warrants may only be exercised for a whole number of shares. No fractional warrants will be issued upon separation of the Units and only whole warrants will trade. The Public Warrants will become exercisable on the later of (a) 30 days after the completion of a Business Combination and (b) 12 months from the closing of the Initial Public Offering. The Public Warrants will expire five years after the completion of a Business Combination or earlier upon redemption or liquidation.
The Company will not be obligated to deliver any shares of Class A common stock pursuant to the exercise of a warrant and will have no obligation to settle such warrant exercise unless a registration statement under the Securities Act with respect to the Class A common stock underlying the warrants is then effective and a prospectus relating thereto is current, subject to the Company satisfying its obligations with respect to
F-19
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
registration. No warrant will be exercisable and the Company will not be obligated to issue shares of Class A common stock upon exercise of a warrant unless the share of Class A common stock issuable upon such warrant exercise has been registered, qualified or deemed to be exempt under the securities laws of the state of residence of the registered holder of the warrants.
The Company has agreed that as soon as practicable, but in no event later than 15 business days, after the closing of a Business Combination, it will use its best efforts to file with the SEC a registration statement covering the shares of Class A common stock issuable upon exercise of the warrants, to cause such registration statement to become effective within 60 business days following a Business Combination and to maintain a current prospectus relating to those shares of Class A common stock until the warrants expire or are redeemed, as specified in the warrant agreement. If a registration statement covering the shares of Class A common stock issuable upon exercise of the warrants is not effective by the 60
th
business day after the closing of a Business Combination, warrant holders may, until such time as there is an effective registration statement and during any period when the Company will have failed to maintain an effective registration statement, exercise warrants on a “cashless basis” in accordance with Section 3(a)(9) of the Securities Act or another exemption. Notwithstanding the above, if the Class A common stock is at the time of any exercise of a warrant not listed on a national securities exchange such that it satisfies the definition of a “covered security” under Section 18(b)(1) of the Securities Act, the Company may, at its option, require holders of Public Warrants who exercise their warrants to do so on a “cashless basis” in accordance with Section 3(a)(9) of the Securities Act and, in the event the Company so elects, the Company will not be required to file or maintain in effect a registration statement, and in the event the Company does not so elect, the Company will use its best efforts to register or qualify the shares under applicable blue sky laws to the extent an exemption is not available.Once the warrants become exercisable, the Company may redeem the outstanding Public Warrants:
| • | in whole and not in part; |
| • | at a price of $0.01 per Public Warrant; |
| • | upon not less than 30 days’ prior written notice of redemption to each warrant holder; and |
| • | if, and only if, the reported last sale price of the Class A common stock equals or exceeds $18.00 per share (as adjusted for stock splits, stock dividends, reorganizations, recapitalizations and the like) for any 20 trading days within a 30-trading day period ending three business trading days before sending the notice of redemption to warrant holders. |
If and when the warrants become redeemable by the Company, the Company may exercise its redemption right even if it is unable to register or qualify the underlying securities for sale under all applicable state securities laws.
If the Company calls the Public Warrants for redemption, management will have the option to require all holders that wish to exercise the Public Warrants to do so on a “cashless basis,” as described in the warrant agreement. The exercise price and number of shares of Class A common stock issuable upon exercise of the warrants may be adjusted in certain circumstances including in the event of a stock dividend, or recapitalization, reorganization, merger or consolidation. However, except as described below, the warrants will not be adjusted for issuance of Class A common stock at a price below its exercise price. Additionally, in no event will the Company be required to net cash settle the warrants. If the Company is unable to complete a Business Combination within the Combination Period and the Company liquidates the funds held in the Trust Account, holders of warrants will not receive any of such funds with respect to their warrants, nor will they receive any distribution from the Company’s assets held outside of the Trust Account with the respect to such warrants. Accordingly, the warrants may expire worthless.
F-20
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
In addition, if (x) the Company issues additional shares of Class A common stock or equity-linked securities for capital raising purposes in connection with the closing of a Business Combination at an issue price or effective issue price of less than $9.20 per share of Class A common stock (with such issue price or effective issue price to be determined in good faith by the Company’s board of directors, and, in the case of any such issuance to the Sponsor or its affiliates, without taking into account any Founder Shares held by the Sponsor or its affiliates, as applicable, prior to such issuance) (the “Newly Issued Price”), (y) the aggregate gross proceeds from such issuances represent more than 60% of the total equity proceeds, and interest thereon, available for the funding of a Business Combination on the date of the completion of a Business Combination (net of redemptions), and (z) the volume weighted average trading price of the Company’s Class A common stock during the 20 trading day period starting on the trading day after the day on which the Company completes a Business Combination (such price, the “Market Value”) is below $9.20 per share, the exercise price of the warrants will be adjusted (to the nearest cent) to be equal to 115% of the higher of the Market Value and the Newly Issued Price, the $18.00 per share redemption trigger price will be adjusted (to the nearest cent) to be equal to 180% of the higher of the Market Value and the Newly Issued Price
.
As of December 31, 2021, there were 2,000,000 Private Placement Warrants outstanding and 750,000 Insider Warrants outstanding which are identical to the Private Placement Warrants. As of December 31, 2020 there were no Private Placement Warrants or Insider Warrants outstanding. The Private Placement Warrants are identical to the Public Warrants underlying the Units sold in the Initial Public Offering, except that (1) the Private Placement Warrants and the Class A common stock issuable upon the exercise of the Private Placement Warrants will not be transferable, assignable or saleable until 30 days after the completion of a Business Combination, subject to certain limited exceptions, (2) the Private Placement Warrants will be exercisable on a cashless basis, (3) the Private Placement Warrants will be
non-redeemable
so long as they are held by the initial purchasers or their permitted transferees, and (4) the holders of the Private Placement Warrants and the Class A common stock issuable upon the exercise of the Private Placement Warrants will have certain registration rights. If the Private Placement Warrants are held by someone other than the initial purchasers or their permitted transferees, the Private Placement Warrants will be redeemable by the Company and exercisable by such holders on the same basis as the Public Warrants.NOTE 9. INCOME TAX
The Company’s net deferred tax assets at December 31, 2021 and 2020 is as follows:
December 31, 2021 | December 31, 2020 | |||||||
Deferred tax asset (liability) | ||||||||
Net operating loss carryforward | $ | 39,939 | $ | 531 | ||||
Startup/Organization Expenses | 202,776 | — | ||||||
Business combination expenses | 2,494,698 | — | ||||||
Total deferred tax assets, net | 2,737,413 | 531 | ||||||
Valuation Allowance | (2,737,413 | ) | (531 | ) | ||||
Deferred tax liability, net of valuation allowance | $ | 0— | $ | 0— | ||||
F-21
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
The income tax provision for the year ended December 31, 2021 and for the period from July 2, 2020 (inception) through December 31, 2020 consists of the following:
December 31, 2021 | December 31, 2020 | |||||||
Federal | ||||||||
Current | $ | 0— | $ | 0— | ||||
Deferred | (2,736,882 | ) | (531 | ) | ||||
State and Local | ||||||||
Current | 0— | 0— | ||||||
Deferred | 0— | 0— | ||||||
Change in valuation allowance | 2,736,882 | 531 | ||||||
Income tax provision | $ | 0— | $ | 0— | ||||
As of December 31, 2021 and 2020, the Company had $187,659 and $2,528 of U.S. federal net operating loss carryovers available to offset future taxable income.
In assessing the realization of the deferred tax assets, management considers whether it is more likely than not that some portion of all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which temporary differences representing net future deductible amounts become deductible. Management considers the scheduled reversal of deferred tax liabilities, projected future taxable income and tax planning strategies in making this assessment. After consideration of all of the information available, management believes that significant uncertainty exists with respect to future realization of the deferred tax assets and has therefore established a full valuation allowance. For the year ended December 31, 2021 and for the period from July 2, 2020 (inception) through December 31, 2020, the change in the valuation allowance was $2,736,882 and $531, respectively.
A reconciliation of the federal income tax rate to the Company’s effective tax rate at December 31, 2021 and 2020 is as follows:
December 31, 2021 | December 31, 2020 | |||||||
Statutory federal income tax rate | 21.0 | % | 21.0 | % | ||||
State taxes, net of federal tax benefit | 0.0 | % | 0.0 | % | ||||
Change in fair value of warrants | (0.98 | )% | 0.0 | % | ||||
Transaction costs allocated to warrants | (0.07 | )% | 0.0 | % | ||||
Fair value of private warrant liability in excess of proceeds | (1.78 | )% | 0.0 | % | ||||
Change in valuation allowance | (18.17 | )% | (21.0 | )% | ||||
Income tax provision | 0.0 | % | 0.0 | % | ||||
The Company files income tax returns in the U.S. federal jurisdiction and is subject to examination by the various taxing authorities. The Company’s tax returns since inception remain open to examination by the taxing authorities.
NOTE 10 — FAIR VALUE MEASUREMENTS
The fair value of the Company’s financial assets and liabilities reflects management’s estimate of amounts that the Company would have received in connection with the sale of the assets or paid in connection with the
F-22
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
transfer of the liabilities in an orderly transaction between market participants at the measurement date. In connection with measuring the fair value of its assets and liabilities, the Company seeks to maximize the use of observable inputs (market data obtained from independent sources) and to minimize the use of unobservable inputs (internal assumptions about how market participants would price assets and liabilities). The following fair value hierarchy is used to classify assets and liabilities based on the observable inputs and unobservable inputs used in order to value the assets and liabilities:
| Level 1: | Quoted prices in active markets for identical assets or liabilities. An active market for an asset or liability is a market in which transactions for the asset or liability occur with sufficient frequency and volume to provide pricing information on an ongoing basis. | |||
| Level 2: | Observable inputs other than Level 1 inputs. Examples of Level 2 inputs include quoted prices in active markets for similar assets or liabilities and quoted prices for identical assets or liabilities in markets that are not active. | |||
| Level 3: | Unobservable inputs based on our assessment of the assumptions that market participants would use in pricing the asset or liability. | |||
The Company classifies its U.S. Treasury and equivalent securities asin accordance with ASC Topic 320 “Investments - Debt and Equity Securities.”securities are those securities which the Company has the ability and intent to hold until maturity.treasury securities are recorded at amortized cost on the accompanying balance sheets and adjusted for the amortization or accretion of premiums or discounts.
held-to-maturity
Held-to-maturity
Held-to-maturity
At December 31, 2021, assets held in the Trust Account were comprised of $207,012,391 in
cash and
money market funds, which primarily invest in U.S. Treasury securities. During the year ended December 31, 2021, the Company did not withdraw any interest income from the Trust Account.The following table presents information about the Company’s assets and liabilities that are measured at fair value under ASC Topic 820, “Fair Value Measurement,” on a recurring basis at December 31, 2021 and indicates the fair value hierarchy of the valuation inputs the Company utilized to determine such fair value.
Level | Fair Value | |||||||
Assets: | ||||||||
Investments held in Trust Account – Cash and money market funds | 1 | $ | 207,012,391 | |||||
Liabilities: | ||||||||
Warrant Liability – Public Warrants | 1 | 12,420,000 | ||||||
Warrant Liability – Private Placement Warrants | 3 | 2,520,000 | ||||||
Warrant Liability - Sponsor and Directors | 3 | 945,000 | ||||||
The Warrants are accounted for as liabilities in accordance with ASC
815-40
and presented within warrant liabilities in the accompanying consolidated balance sheets. The warrant liabilities are measured at fair value at inception and on a recurring basis, with changes in fair value presented within change in fair value of warrant liabilities in the consolidated statements of operations.The Private Placement Warrants were initially valued using a Modified Black Scholes Option Pricing Model, which is considered to be a Level 3 fair value measurement. The Modified Black Scholes model’s primary unobservable input utilized in determining the fair value of the Private Placement Warrants is the expected volatility of the common stock. The expected volatility as of the Initial Public Offering date was
F-23
GREENLIGHT BIOSCIENCES HOLDINGS, PBC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
DECEMBER 31, 2021
derived from observable public warrant pricing on comparable ‘blank-check’ companies without an identified target. The expected volatility as of subsequent valuation dates was implied from the Company’s own Public Warrant pricing. A Monte Carlo simulation methodology was used in estimating the fair value of the Public Warrants for periods where no observable traded price was available, using the same expected volatility as was used in measuring the fair value of the Private Placement Warrants. For periods subsequent to the detachment of the warrants from the Units, the close price of the Public Warrant price was used as the fair value as of each relevant date. The measurement of the Public Warrants after the detachment of the Public Warrants from the Units is classified as Level 1 due to the use of an observable market quote in an active market.
The key inputs into the Black-Scholes-Merton model for the warrants were as follows:
Input | January 13, 2021 | December 31, 2021 | ||||||
Risk-free interest rate | 0.74 | % | 1.30 | % | ||||
Expected term (years) | 5.00 | 5.00 | ||||||
Expected volatility | 21 | % | 18.0 | % | ||||
Exercise price | $ | 11.50 | $ | 11.50 | ||||
Fair value of Units | $ | 9.43 | $ | 9.92 | ||||
The following table presents the changes in the fair value of Level 3 warrant liabilities:
Private Placement | Public | Warrant Liabilities | ||||||||||
Fair value as of January 1, 2021 | $ | 0 | $ | 0 | $ | 0 | ||||||
Initial measurement on January 19, 2021 | 3,272,500 | 11,902,500 | 15,175,000 | |||||||||
Change in valuation inputs or other assumptions | 192,500 | (2,898,000 | ) | (2,705,500 | ) | |||||||
Transfers to Level 1 | 0 | (9,004,500 | ) | (9,004,500 | ) | |||||||
Fair value as of December 31, 2021 | $ | 3,465,000 | $ | 0 | $ | 3,465,000 | ||||||
Transfers to/from Levels 1, 2 and 3 are recognized at the end of the reporting period in which a change in valuation technique or methodology occurs. The estimated fair value of the Public Warrants transferred from a Level 3 measurement to a Level 1 fair value measurement during the year ended December 31, 2021 was approximately $9.0 million, when the Public Warrants were separately listed and traded.
NOTE 11 — SUBSEQUENT EVENTS
The Company evaluated subsequent events and transactions that occurred after the balance sheet date up to the date that the financial statements were issued. Other than the business combination as discussed in Note 1 and Note 6, the Company did not identify any subsequent events that would have required adjustment or disclosure in the
consolidated
financial statements.F-2
4